











-
Organic-inorganic halide perovskites ABX3 (A = CH3NH3, HC(NH2)2; B = Pb; X = Cl, Br, I) have recently attracted increasing attention due to their advanced optoelectronic properties. However, the poor stability and toxicity of organic lead halogen perovskites are still a major challenge for deploying the outdoor solar cells. Element substitution is a simple and effective strategy to solve these problems. For example, the substitution of the I ions with Cl and Br has been regarded as a reliable method to improve the device stability. A-site engineering, i.e., replacing organic ions with inorganic cations (such as Cs+, Rb+), has also been reported. The B-site alloying approach has been demonstrated with Zn, Sr, Sn, etc. Inorganic halide perovskites can be synthesized by the low-cost solution spin-coating method and have similar optoelectronic properties and improved stability to their organic counterparts. Here in this paper, we report a comprehensive study of the alloyed perovskite CsPb1–xBaxX3 (X = Cl, Br, I) by combining the disorder alloy structure search method with first-principles energy calculations. We find that it is not easy to dope barium into the perovskite lattice when Ba concentration is low and the stable disordered solid solution can exist in the high Ba concentration case. Carrier effective mass and bandgap increase with the increase of Ba concentration and the bandgap change range is wide, owing to the difference in both electronegativity and ionic radius between Pb and Ba. After inducing Ba into CsPb1–xBaxX3 (X = Cl, Br, I), the higher electron concentration on the I sites also enhances the Coulomb interaction of the Pb—I bonds. Moreover, the electrons and holes tend to be located on Pb sites, and this may give rise to the formation of local potential wells, which would further induce the large lattice deformation to accommodate the self-trapped excitons. Especially, CsPbI3-Pnma perovskite is metastable in the ambient environment with a suitable photon absorption threshold. The CsPb1–xBaxI3 can be used as a capping layer on CsPbI3 in solar cells, thereby significantly improving the power conversion efficiency and long-term stability. Overall, the alloyed perovskite CsPb1–xBaxX3 (X = Cl, Br, I) with high Ba concentration can be stable and less-toxic, and they can be used in short wave light-emitting diodes, radiation detectors or other fields because of their large bandgaps (> 2.8 eV).
-
Keywords:
- lead-based halide perovskite /
- elemental substitution /
- optoelectronic properties /
- first-principles calculations
[1] Bell L E 2008 Science 321 1457
 Google Scholar
Google Scholar
[2] Zou C, Zhao Q, Zhang G, Xiong B 2016 Natural Gas Industry B 3 1
Google Scholar
[3] Lenzen M 2008 Energy Conversion and Management 49 2178
Google Scholar
[4] Polman A, Knight M, Garnett E C, Ehrler B, Sinke W C 2016 Science 352 aad4424
Google Scholar
[5] Milan P, Wächter M, Peinke J 2013 Phys. Rev. Lett. 110 138701
Google Scholar
[6] Lewis N S 2007 Science 315 798
Google Scholar
[7] Dong Q, Fang Y, Shao Y, Mulligan P, Qiu J, Cao L, Huang J 2015 Science 347 967
Google Scholar
[8] Xing G, Mathews N, Sun S, Lim S S, Lam Y M, Gratzel M, Mhaisalkar S, Sum T C 2013 Science 342 344
Google Scholar
[9] Han Q, Bae S H, Sun P, Hsieh Y T, Yang Y M, Rim Y S, Zhao H, Chen Q, Shi W, Li G, Yang Y 2016 Adv. Mater. 28 2253
Google Scholar
[10] Chen H, Xiang S, Li W, Liu H, Zhu L, Yang S 2018 Solar RRL 2 1700188
Google Scholar
[11] Shang M H, Zhang J, Zhang P, Yang Z, Zheng J, Haque M A, Yang W, Wei S H, Wu T 2019 J. Phys. Chem. Lett. 10 59
Google Scholar
[12] Huang, Y, Sun Q D, Xu W, He Y, Yin W J 2017 Acta Phys.-Chim. Sin. 33 1730
[13] Qin X, Zhao Z, Wang Y, Wu J, Jiang Q, You J 2017 J. Semicond. 38 011002
Google Scholar
[14] Linaburg M R, McClure E T, Majher J D, Woodward P M 2017 Chem. Mater. 29 3507
Google Scholar
[15] Wu M C, Chen W C, Chan S H, Su W F 2018 Appl. Surf. Sci. 429 9
Google Scholar
[16] Lau C F J, Zhang M, Deng X, Zheng J, Bing J, Ma Q, Kim J, Hu L, Green M A, Huang S, Ho-Baillie A 2017 ACS Energy Lett. 2 2319
Google Scholar
[17] Navas J, Sánchez-Coronilla A, Gallardo J J, Cruz Hernández N, Piñero J C, Alcántara R, Fernández-Lorenzo C, De los Santos D M, Aguilar T, Martín-Calleja J 2015 Nanoscale 7 6216
Google Scholar
[18] Li F, Xia Z, Gong Y, Gu L, Liu Q 2017 J. Mater. Chem. C 5 9281
Google Scholar
[19] Bechtel J S, van der Ven A 2018 Phys. Rev. Mater. 2 045401
Google Scholar
[20] Fu Y, Rea M T, Chen J, Morrow D J, Hautzinger M P, Zhao Y, Pan D, Manger L H, Wright J C, Goldsmith R H, Jin S 2017 Chem. Mater. 29 8385
Google Scholar
[21] Wang P, Zhang X, Zhou Y, Jiang Q, Ye Q, Chu Z, Li X, Yang X, Yin Z, You J 2018 Nat. Commun. 9 2225
Google Scholar
[22] Ju M G, Dai J, Ma L, Zeng X C 2017 J. Am. Chem. Soc. 139 8038
Google Scholar
[23] Hao F, Stoumpos C C, Cao D H, Chang R P H, Kanatzidis M G 2014 Nature Photonics 8 489
Google Scholar
[24] Swarnkar A, Mir W J, Nag A 2018 ACS Energy Lett. 3 286
Google Scholar
[25] Xiang W, Wang Z, Kubicki D J, Tress W, Luo J, Prochowicz D, Akin S, Emsley L, Zhou J, Dietler G, Grätzel M, Hagfeldt A 2019 Joule 3 205
Google Scholar
[26] Pazoki M, Jacobsson T J, Hagfeldt A, Boschloo G, Edvinsson T 2016 Phys. Rev. B 93 144105
Google Scholar
[27] Huang Q, Zou Y, Bourelle S A, Zhai T, Wu T, Tan Y, Li Y, Li J, Duhm S, Song T, Wang L, Deschler F, Sun B 2019 Nanoscale Horizons DOI: 10.1039.C9NH00066F
[28] Kumar A, Balasubramaniam K R, Kangsabanik J, Vikram, Alam A 2016 Phys. Rev. B 94 180105
Google Scholar
[29] Song J, Li J, Li X, Xu L, Dong Y, Zeng H 2015 Adv. Mater. 27 7162
Google Scholar
[30] Li X, Wu Y, Zhang S, Cai B, Gu Y, Song J, Zeng H 2016 Adv. Funct. Mater. 26 2435
Google Scholar
[31] van de Walle A, Tiwary P, de Jong M, Olmsted D L, Asta M, Dick A, Shin D, Wang Y, Chen L Q, Liu Z K 2013 Calphad 42 13
Google Scholar
[32] Hass K C, Davis L C, Zunger A 1990 Phys. Rev. B 42 3757
Google Scholar
[33] Jiang C, Stanek C R, Sickafus K E, Uberuaga B P 2009 Phys. Rev. B 79 104203
Google Scholar
[34] Shin D, van de Walle A, Wang Y, Liu Z K 2007 Phys. Rev. B 76 144204
Google Scholar
[35] Kresse G, Furthmüller J 1996 Phys. Rev. B 54 11169
Google Scholar
[36] Kresse G, Furthmüller J 1996 Comput. Mater. Sci. 6 15
Google Scholar
[37] Grimme S 2006 J. Comput. Chem. 27 1787
Google Scholar
[38] Gajdoš M, Hummer K, Kresse G, Furthmüller J, Bechstedt F 2006 Phys. Rev. B 73 045112
Google Scholar
[39] Hu J, Alicea J, Wu R, Franz M 2012 Phys. Rev. Lett. 109 266801
Google Scholar
[40] Feng Y, Ding H C, Du Y, Wan X, Wang B, Savrasov S Y, Duan C G 2014 J. Appl. Phys. 115 233901
Google Scholar
[41] Yun S, Zhou X, Even J, Hagfeldt A 2017 Angew. Chem. Int. Ed. 56 15806
Google Scholar
[42] Krukau A V, Vydrov O A, Izmaylov A F, Scuseria G E 2006 J. Chem. Phys. 125 224106
Google Scholar
[43] Perdew J P, Burke K, Ernzerhof M 1996 Phys. Rev. Lett. 77 3865
Google Scholar
[44] Medeiros P V C, Stafström S, Björk J 2014 Phys. Rev. B 89 041407
Google Scholar
[45] Medeiros P V C, Tsirkin S S, Stafström S, Björk J 2015 Phys. Rev. B 91 041116
Google Scholar
[46] Pauling L 1932 J. Am. Chem. Soc. 54 3570
Google Scholar
[47] Yu J, Kong J, Hao W, Guo X, He H, Leow W R, Liu Z, Cai P, Qian G, Li S, Chen X, Chen X 2019 Adv. Mater. 31 1806385
[48] Tanaka K, Kondo T 2003 Sci. Technol. Adv. Mater. 4 599
Google Scholar
[49] Lee K J, Turedi B, Sinatra L, Zhumekenov A A, Maity P, Dursun I, Naphade R, Merdad N, Alsalloum A, Oh S, Wehbe N, Hedhili M N, Kang C H, Subedi R C, Cho N, Kim J S, Ooi B S, Mohammed O F, Bakr O M 2019 Nano Lett. 19 3535
[50] Jiang Y, Qin C, Cui M, He T, Liu K, Huang Y, Luo M, Zhang L, Xu H, Wei J, Liu Z, Wang H, Kim G H, Yuan M, Chen J 2019 Nat. Commun. 10 1868
Google Scholar
[51] Zhang S, Yi C, Wang N, Sun Y, Zou W, Wei Y, Cao Y, Miao Y, Li R, Yin Y, Zhao N, Wang J, Huang W 2017 Adv. Mater. 29 1606600
Google Scholar
[52] Blancon J C, Stier A V, Tsai H, Nie W, Stoumpos C C, Traoré B, Pedesseau L, Kepenekian M, Katsutani F, Noe G T, Kono J, Tretiak S, Crooker S A, Katan C, Kanatzidis M G, Crochet J J, Evan J, Mohite A D 2018 Nat. Commun. 9 2254
Google Scholar
[53] Kulbak M, Cahen D, Hodes G 2015 J. Phys. Chem. Lett. 6 2452
Google Scholar
-
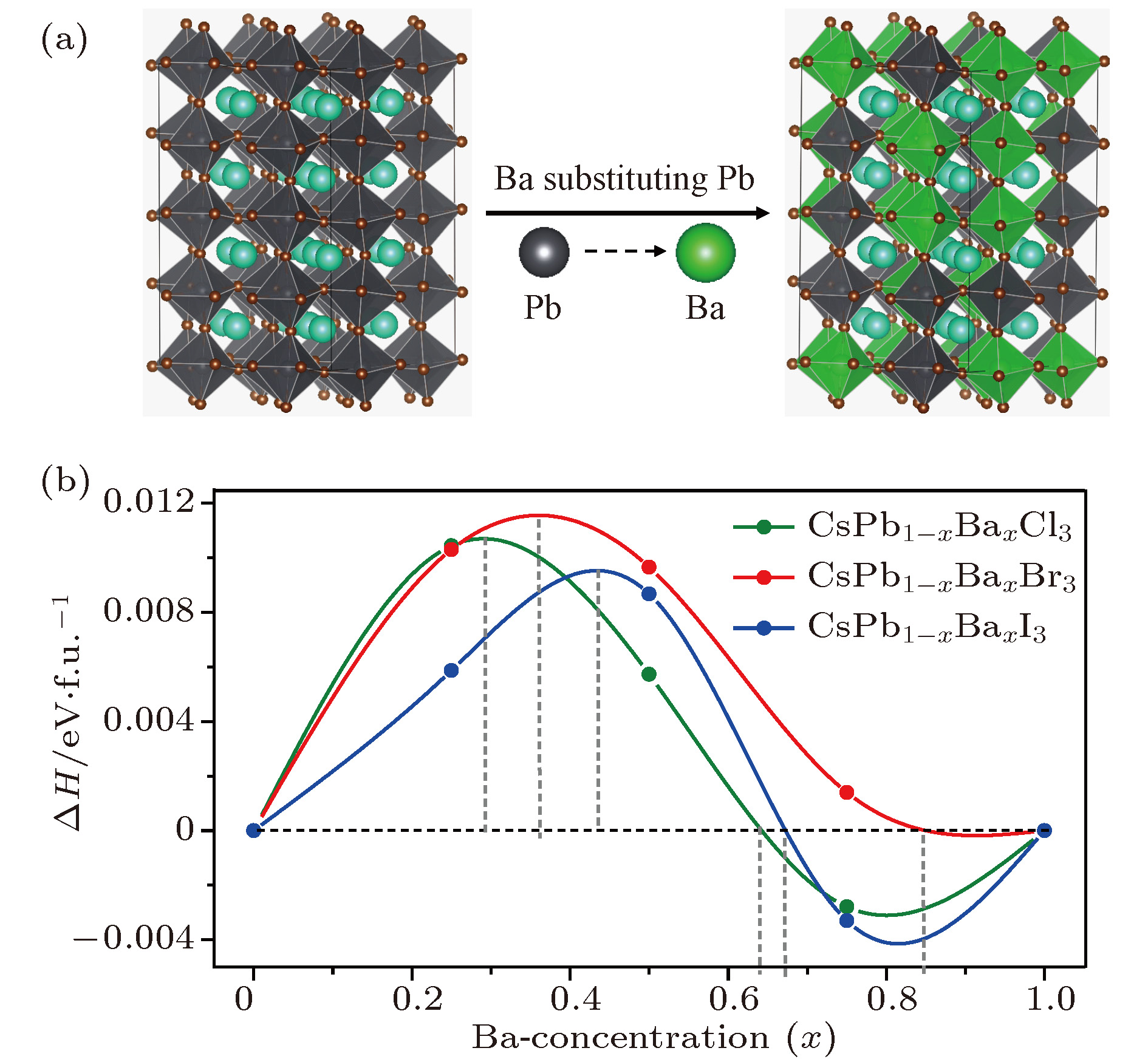
图 1 CsPb1-xBaxX3 (X = Cl, Br, I; x = 0, 0.25, 0.5, 0.75, 1)合金钙钛矿体系的(a)晶体结构示意图, (b)计算得到的形成能
Figure 1. (a) Schematic diagram of crystal structure, (b) density functional theory-calculated formation energies of the alloyed perovskite CsPb1-xBaxX3 (X = Cl, Br, I; x = 0, 0.25, 0.5, 0.75, 1).
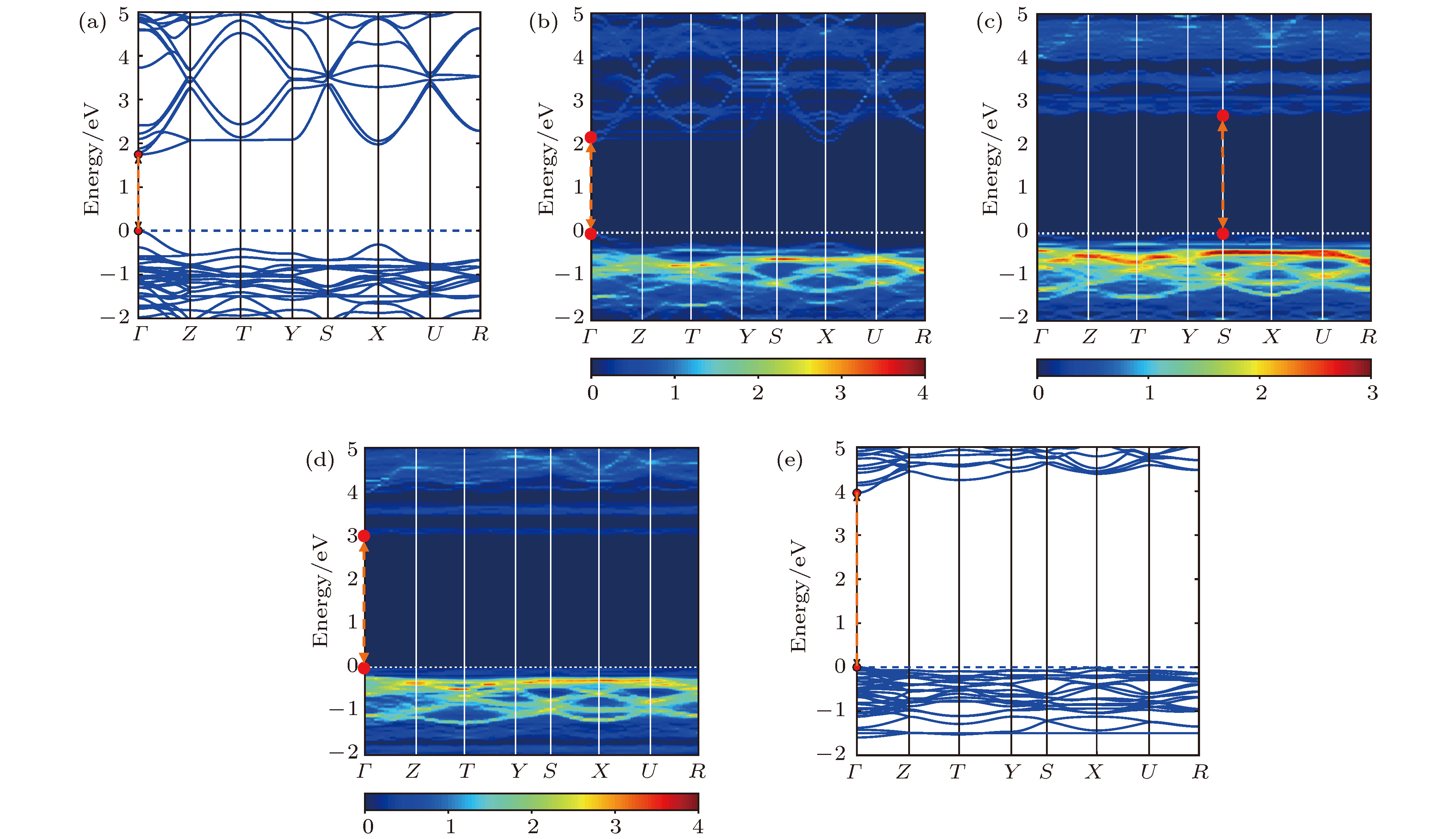
图 2 计算得到的CsPb1–xBaxI3合金钙钛矿体系的能带结构 (a) x = 0%; (b) x = 25%; (c) x = 50%; (d) x = 75%; (e) x = 100%; 其中, 图(b)−(d)是通过能带展开技术得到的, 彩色刻度尺代表指定波矢下穿过能量区间的原胞能带数目
Figure 2. Calculated band structures of the alloyed perovskite CsPb1–xBaxI3, x = (a) 0%, (b) 25%, (c) 50%, (d) 75%, (e) 100%; panels (b)−(d) are obtained by band unfolding technique. The color scale represents the number of the primitive cell bands crossing the energy interval at a given primitive wave vector.
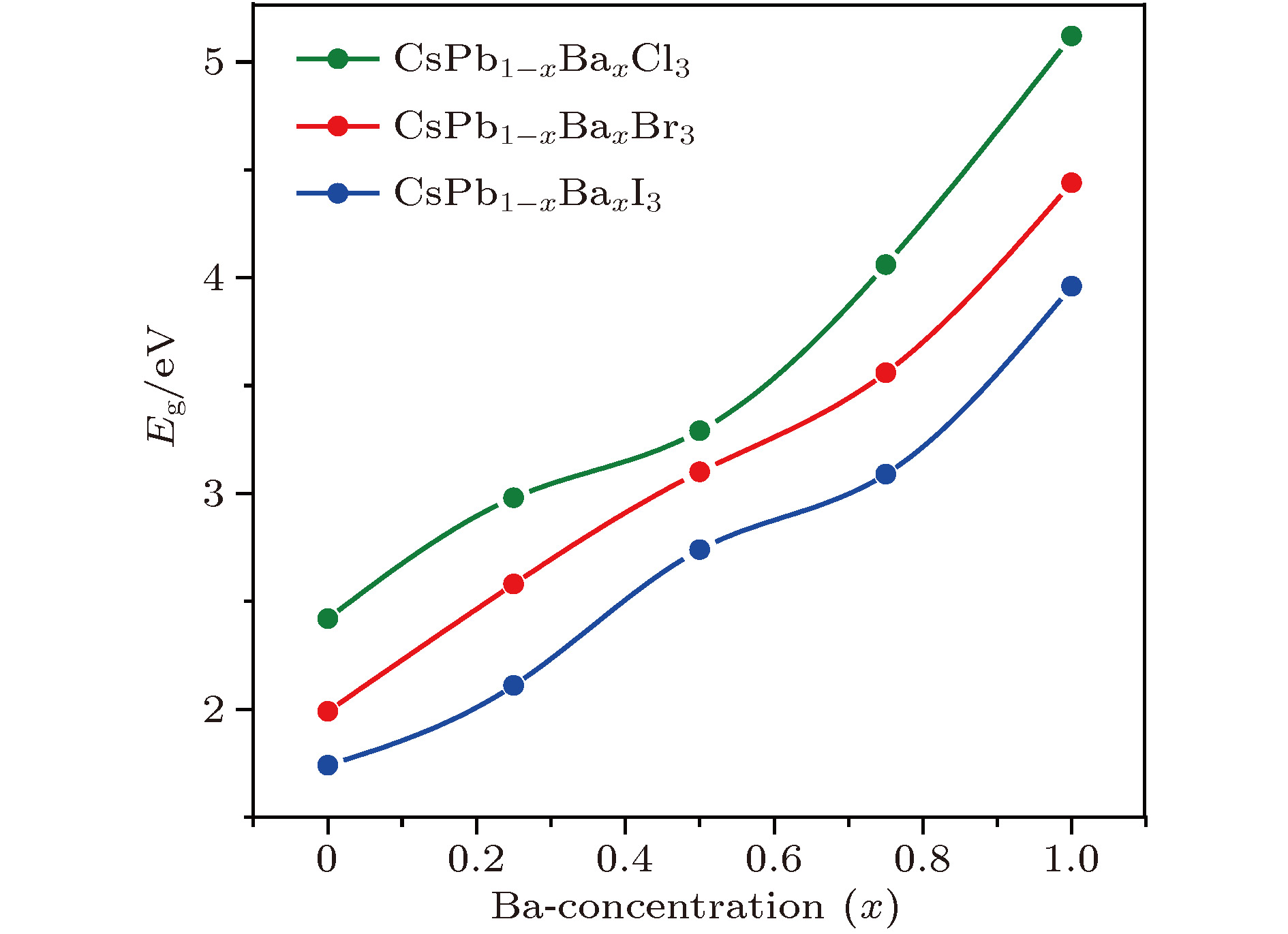
图 3 计算得到的CsPb1–xBaxX3 (X = Cl, Br, I; x = 0, 0.25, 0.5, 0.75, 1)合金钙钛矿体系的带隙变化规律
Figure 3. Calculated band gaps of the alloyed perovskite CsPb1–xBaxX3 (X = Cl, Br, I; x = 0, 0.25, 0.5, 0.75, 1) varied with Ba concentration.
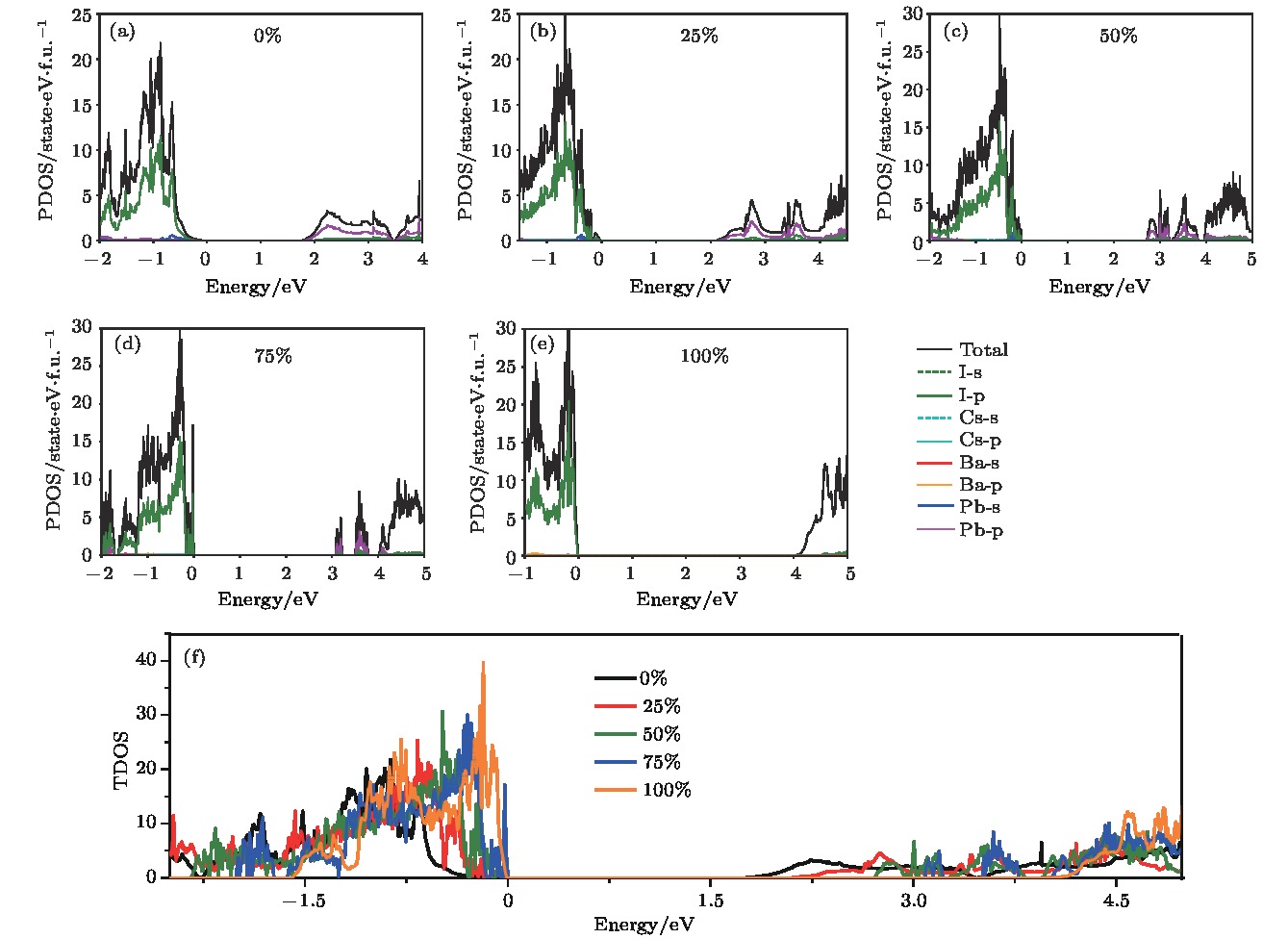
图 4 计算得到的CsPb1–xBaxI3 (x = 0, 0.25, 0.5, 0.75, 1)合金钙钛矿体系的(a)−(e)投影态密度, (f)总态密度
Figure 4. Calculated (a)−(e) Atomic-orbital-projected density of states (PDOS), (f) total density of states (TDOS) of the alloyed perovskite CsPb1–xBaxI3 (x = 0, 0.25, 0.5, 0.75, 1).

图 5 计算得到的CsPb1-xBaxI3合金钙钛矿体系的部分电荷密度分布图样 (a), (f) 0%; (b), (g) 25%; (c), (h) 50%; (d), (i) 75%; (e), (j) 100%
Figure 5. Calculated partial charge distribution patterns of the alloyed perovskite CsPb1-xBaxI3: (a), (f) 0%; (b), (g) 25%; (c), (h) 50%; (d), (i) 75%; (e), (j) 100%.
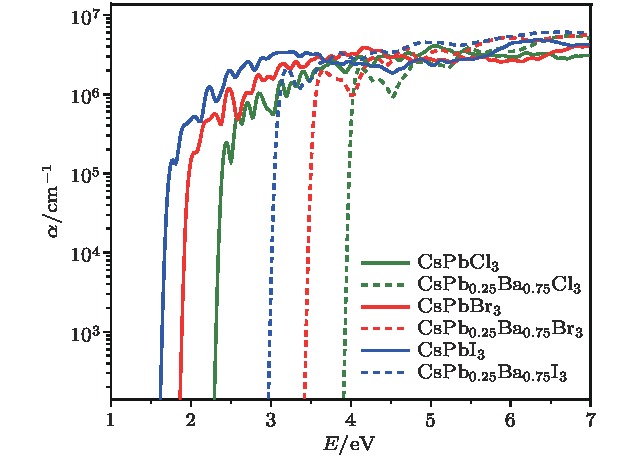
图 6 计算得到的CsPbX3和CsPb0.25Ba0.75X3 (X = Cl, Br, I)合金钙钛矿的光吸收谱
Figure 6. Calculated photo absorption spectra of the perovskite CsPbX3 and CsPb0.25Ba0.75X3 (X = Cl, Br, I).
-
[1] Bell L E 2008 Science 321 1457
Google Scholar
[2] Zou C, Zhao Q, Zhang G, Xiong B 2016 Natural Gas Industry B 3 1
Google Scholar
[3] Lenzen M 2008 Energy Conversion and Management 49 2178
Google Scholar
[4] Polman A, Knight M, Garnett E C, Ehrler B, Sinke W C 2016 Science 352 aad4424
Google Scholar
[5] Milan P, Wächter M, Peinke J 2013 Phys. Rev. Lett. 110 138701
Google Scholar
[6] Lewis N S 2007 Science 315 798
Google Scholar
[7] Dong Q, Fang Y, Shao Y, Mulligan P, Qiu J, Cao L, Huang J 2015 Science 347 967
Google Scholar
[8] Xing G, Mathews N, Sun S, Lim S S, Lam Y M, Gratzel M, Mhaisalkar S, Sum T C 2013 Science 342 344
Google Scholar
[9] Han Q, Bae S H, Sun P, Hsieh Y T, Yang Y M, Rim Y S, Zhao H, Chen Q, Shi W, Li G, Yang Y 2016 Adv. Mater. 28 2253
Google Scholar
[10] Chen H, Xiang S, Li W, Liu H, Zhu L, Yang S 2018 Solar RRL 2 1700188
Google Scholar
[11] Shang M H, Zhang J, Zhang P, Yang Z, Zheng J, Haque M A, Yang W, Wei S H, Wu T 2019 J. Phys. Chem. Lett. 10 59
Google Scholar
[12] Huang, Y, Sun Q D, Xu W, He Y, Yin W J 2017 Acta Phys.-Chim. Sin. 33 1730
[13] Qin X, Zhao Z, Wang Y, Wu J, Jiang Q, You J 2017 J. Semicond. 38 011002
Google Scholar
[14] Linaburg M R, McClure E T, Majher J D, Woodward P M 2017 Chem. Mater. 29 3507
Google Scholar
[15] Wu M C, Chen W C, Chan S H, Su W F 2018 Appl. Surf. Sci. 429 9
Google Scholar
[16] Lau C F J, Zhang M, Deng X, Zheng J, Bing J, Ma Q, Kim J, Hu L, Green M A, Huang S, Ho-Baillie A 2017 ACS Energy Lett. 2 2319
Google Scholar
[17] Navas J, Sánchez-Coronilla A, Gallardo J J, Cruz Hernández N, Piñero J C, Alcántara R, Fernández-Lorenzo C, De los Santos D M, Aguilar T, Martín-Calleja J 2015 Nanoscale 7 6216
Google Scholar
[18] Li F, Xia Z, Gong Y, Gu L, Liu Q 2017 J. Mater. Chem. C 5 9281
Google Scholar
[19] Bechtel J S, van der Ven A 2018 Phys. Rev. Mater. 2 045401
Google Scholar
[20] Fu Y, Rea M T, Chen J, Morrow D J, Hautzinger M P, Zhao Y, Pan D, Manger L H, Wright J C, Goldsmith R H, Jin S 2017 Chem. Mater. 29 8385
Google Scholar
[21] Wang P, Zhang X, Zhou Y, Jiang Q, Ye Q, Chu Z, Li X, Yang X, Yin Z, You J 2018 Nat. Commun. 9 2225
Google Scholar
[22] Ju M G, Dai J, Ma L, Zeng X C 2017 J. Am. Chem. Soc. 139 8038
Google Scholar
[23] Hao F, Stoumpos C C, Cao D H, Chang R P H, Kanatzidis M G 2014 Nature Photonics 8 489
Google Scholar
[24] Swarnkar A, Mir W J, Nag A 2018 ACS Energy Lett. 3 286
Google Scholar
[25] Xiang W, Wang Z, Kubicki D J, Tress W, Luo J, Prochowicz D, Akin S, Emsley L, Zhou J, Dietler G, Grätzel M, Hagfeldt A 2019 Joule 3 205
Google Scholar
[26] Pazoki M, Jacobsson T J, Hagfeldt A, Boschloo G, Edvinsson T 2016 Phys. Rev. B 93 144105
Google Scholar
[27] Huang Q, Zou Y, Bourelle S A, Zhai T, Wu T, Tan Y, Li Y, Li J, Duhm S, Song T, Wang L, Deschler F, Sun B 2019 Nanoscale Horizons DOI: 10.1039.C9NH00066F
[28] Kumar A, Balasubramaniam K R, Kangsabanik J, Vikram, Alam A 2016 Phys. Rev. B 94 180105
Google Scholar
[29] Song J, Li J, Li X, Xu L, Dong Y, Zeng H 2015 Adv. Mater. 27 7162
Google Scholar
[30] Li X, Wu Y, Zhang S, Cai B, Gu Y, Song J, Zeng H 2016 Adv. Funct. Mater. 26 2435
Google Scholar
[31] van de Walle A, Tiwary P, de Jong M, Olmsted D L, Asta M, Dick A, Shin D, Wang Y, Chen L Q, Liu Z K 2013 Calphad 42 13
Google Scholar
[32] Hass K C, Davis L C, Zunger A 1990 Phys. Rev. B 42 3757
Google Scholar
[33] Jiang C, Stanek C R, Sickafus K E, Uberuaga B P 2009 Phys. Rev. B 79 104203
Google Scholar
[34] Shin D, van de Walle A, Wang Y, Liu Z K 2007 Phys. Rev. B 76 144204
Google Scholar
[35] Kresse G, Furthmüller J 1996 Phys. Rev. B 54 11169
Google Scholar
[36] Kresse G, Furthmüller J 1996 Comput. Mater. Sci. 6 15
Google Scholar
[37] Grimme S 2006 J. Comput. Chem. 27 1787
Google Scholar
[38] Gajdoš M, Hummer K, Kresse G, Furthmüller J, Bechstedt F 2006 Phys. Rev. B 73 045112
Google Scholar
[39] Hu J, Alicea J, Wu R, Franz M 2012 Phys. Rev. Lett. 109 266801
Google Scholar
[40] Feng Y, Ding H C, Du Y, Wan X, Wang B, Savrasov S Y, Duan C G 2014 J. Appl. Phys. 115 233901
Google Scholar
[41] Yun S, Zhou X, Even J, Hagfeldt A 2017 Angew. Chem. Int. Ed. 56 15806
Google Scholar
[42] Krukau A V, Vydrov O A, Izmaylov A F, Scuseria G E 2006 J. Chem. Phys. 125 224106
Google Scholar
[43] Perdew J P, Burke K, Ernzerhof M 1996 Phys. Rev. Lett. 77 3865
Google Scholar
[44] Medeiros P V C, Stafström S, Björk J 2014 Phys. Rev. B 89 041407
Google Scholar
[45] Medeiros P V C, Tsirkin S S, Stafström S, Björk J 2015 Phys. Rev. B 91 041116
Google Scholar
[46] Pauling L 1932 J. Am. Chem. Soc. 54 3570
Google Scholar
[47] Yu J, Kong J, Hao W, Guo X, He H, Leow W R, Liu Z, Cai P, Qian G, Li S, Chen X, Chen X 2019 Adv. Mater. 31 1806385
[48] Tanaka K, Kondo T 2003 Sci. Technol. Adv. Mater. 4 599
Google Scholar
[49] Lee K J, Turedi B, Sinatra L, Zhumekenov A A, Maity P, Dursun I, Naphade R, Merdad N, Alsalloum A, Oh S, Wehbe N, Hedhili M N, Kang C H, Subedi R C, Cho N, Kim J S, Ooi B S, Mohammed O F, Bakr O M 2019 Nano Lett. 19 3535
[50] Jiang Y, Qin C, Cui M, He T, Liu K, Huang Y, Luo M, Zhang L, Xu H, Wei J, Liu Z, Wang H, Kim G H, Yuan M, Chen J 2019 Nat. Commun. 10 1868
Google Scholar
[51] Zhang S, Yi C, Wang N, Sun Y, Zou W, Wei Y, Cao Y, Miao Y, Li R, Yin Y, Zhao N, Wang J, Huang W 2017 Adv. Mater. 29 1606600
Google Scholar
[52] Blancon J C, Stier A V, Tsai H, Nie W, Stoumpos C C, Traoré B, Pedesseau L, Kepenekian M, Katsutani F, Noe G T, Kono J, Tretiak S, Crooker S A, Katan C, Kanatzidis M G, Crochet J J, Evan J, Mohite A D 2018 Nat. Commun. 9 2254
Google Scholar
[53] Kulbak M, Cahen D, Hodes G 2015 J. Phys. Chem. Lett. 6 2452
Google Scholar
-
 157101-20190596补充材料.pdf
157101-20190596补充材料.pdf




 DownLoad:
DownLoad:
Catalog
Metrics
- Abstract views: 21097
- PDF Downloads: 349
- Cited By: 0
