











-
Ternary metal halides have attracted much attention as a new potential photoelectric material due to their ultra-high photoelectric conversion efficiencies. In this paper, USPEX, a crystal structure prediction software based on genetic algorithm, is used to investigate the potential crystal structures of ternary CuBiI compounds (CuBi2I7, Cu2BiI5, Cu2BiI7,Cu3BiI6, Cu3Bi2I9, CuBi3I10, and Cu4BiI7) at atmospheric pressure and absolute zero temperature. Based on the density functional theory, the formation energies, elastic coefficients, and phonon dispersion curves of the predicted structures are calculated. The twelve stable CuBiI compounds with good thermodynamic, dynamical and mechanical stabilities are identified. The twelve crystal structures of CuBiI compound feature mainly the co-existence of Cu—I and Bi—I bonds and coordination polyhedrons of I atoms. The band gaps of twelve structures, calculated by HSE06 method, are 1.13–3.09 eV, indicating that the stoichiometric ratio affects the band gap obviously. Among them, the band gaps of Cu2BiI5-P1, Cu2BiI7-P1 and Cu2BiI7-P1-II are relatively small, close to the optimal band gap value for light absorption (1.40 eV), demonstrating that these compounds are suitable for serving as light absorbing materials in solar cells. The distribution of density of state (DOS) indicates that the top of the valence band of CuBiI compound is attributed to the hybridized Cu-3d and I-5p orbitals; the bottom of the conduction band of Cu3BiI6-R3 comes mainly from the Bi-6p and I-5p orbitals, and Cu-3d contributes little; the conduction band bottom of Cu2BiI7 is mainly from the I-5p orbital, and the Cu-3d has little contribution. The bottoms of the conduction band of other structures originate mainly from the hybridized Bi-6p and I-5p orbitals. Electronic localization function and Bader charge analysis show that the Cu—I and Bi—I bonds have more ionic features and less covalent natures. The DOS distribution also confirms the covalent interaction of Cu/Bi-I. In addition, the CuBiI ternary compounds have extremely strong light absorption capacities (light absorption coefficient higher than 4 × 105 cm–1) in the high-energy region of visible light and high power conversion efficiency (31.63%), indicating that the CuBiI ternary compounds have the potential to be an excellent photoelectric absorption material. Our investigation suggests the further study and potential applications of CuBiI ternary compound as absorber materials in solar cell.
-
Keywords:
- ternary metal halides /
- first principles /
- crystal structure prediction /
- photoelectric conversion efficiency
[1] Zhang F, Lu H P, Tong J H, Berry J J, Beard M C, Zhu K 2020 Energy Environ. Sci. 13 1154
 Google Scholar
Google Scholar
[2] Ajayan J, Nirmal D, Mohankumar P, Saravanan M, Jagadesh M, Arivazhagan L A 2020 Superlattices Microstruct. 143 106549
Google Scholar
[3] Wang L L, Fan B B, Zheng B, Yang Z B, Yin P G, Huo L J 2020 Sustainable Energy Fuels 4 2134
Google Scholar
[4] Chen W J, Li X Q, Li Y W, Li Y F 2020 Energy Environ. Sci. 13 1971
Google Scholar
[5] Wang Y, Sun H D 2018 Small Methods 21 700252
[6] Xi J, Wu Z X, Jiao B, Dong H, Ran C X, Piao C C, Lei T, Song T B, Ke W J, Yokoyama T, Hou X, Kanatzidis M G 2017 Adv. Mater. 29 1606964
Google Scholar
[7] Cheng P F, Wu T, Zhang J W, Li Y J, Liu J X, Jiang L, Mao X, Lu R F, Deng W Q, Han K L 2017 J. Phys. Chem. Lett. 8 4402
Google Scholar
[8] Sumathi R, Johnson K, Viswanathan B, Varadarajan T K 1998 Appl. Catal., A 172 15
Google Scholar
[9] Zhu H X, Liu J M 2016 Sci. Rep. 6 37425
Google Scholar
[10] Peng L, Xie W 2020 RSC Adv. 10 14679
Google Scholar
[11] Jiao Y Q, Lv Y Y, Li J, Niu M, Yang Z Q 2017 Comput. Theor. Chem. 15 20
[12] Zhang L, Liu C M, Lin Y 2019 J. Phys. Chem. Lett. 10 1676
Google Scholar
[13] Pa J, Bhunia A, Chakraborty S, Manna S, Das S, Dewan A, Datta S, Nag A 2018 J. Phys. Chem. C 122 10643
Google Scholar
[14] Sun S J, Tominaka S, Lee J H, Xie F, Bristowe P D, Cheetham A K 2016 APL Mater. 4 031101
Google Scholar
[15] Lu C J, Zhang J, Sun H R, Hou D G, Gan X L, Shang M H, Li Y Y, Hu Z Y, Zhu Y J, Han L Y 2018 ACS Appl. Energy Mater. 1 4485
Google Scholar
[16] Kulkarni A, Jena A K, Ikegami M 2019 Chem. Commun. 55 4031
Google Scholar
[17] Ramachandrana A A, Krishnana B D, Leal D A A, Martinez E G, Martinez J A A, Avellaneda D A, Shaji S 2020 Mater. Today Commun. 24 101092
Google Scholar
[18] Seo Y, Ha S R, Yoon S, Jeong S M, Choi H, Kang D W 2020 J. Power Sources 453 227903
Google Scholar
[19] Yi Z J, Zhang T, Ban H X, Shao H, Gong X, Wu M A, Liang G J, Zhang X L, Shen Y, Wang M K 2020 Sol. Energy 206 436
Google Scholar
[20] Fourcroy P H, Carré D, Thévet F, Rivet J 1991 Acta Crystallogr., Sect. C: Cryst. Struct. Commun. 47 2023
Google Scholar
[21] Hu Z S, Wang Z, Kapil G, Ma T L, Iikubo S, Minemoto T, Yoshino K, Toyoda T, Shen Q, Hayase S 2018 ChemSusChem 11 2930
Google Scholar
[22] Zhang B S, Lei Y, Qi R J, Yu H L, Yang X G, Cai T, Zheng Z 2019 Sci. China Mater. 62 519
Google Scholar
[23] Bi L Y, Hu Y Q, Li M Q, Hu T L, Zhang H L, Yin X T, Que W X, Lassoued M S, Zheng Y Z 2019 J. Mater. Chem. A. 7 19662
Google Scholar
[24] Lyakhov A O, Oganov A R, Stokes H T, Zhu Q 2013 Comput. Phys. Commun. 184 1172
Google Scholar
[25] Li Y L, Wang S N, Oganov A R, Gou H, Smith J S, Strobel T 2015 Nat. Commun. 6 6974
Google Scholar
[26] Kresse G, Furthmüller J 1996 Phys. Rev. B 54 11169
Google Scholar
[27] Perdew J P, Burke K, Ernzerhof M 1996 Phys. Rev. Lett. 77 3865
Google Scholar
[28] Blöchl P E 1994 Phys. Rev. B 50 17953
Google Scholar
[29] Monkhorst H J, Pack J D 1976 Phys. Rev. B 13 5188
Google Scholar
[30] Parlinski K, Li Z Q, Kawazoe Y 1997 Phys. Rev. Lett. 78 4063
Google Scholar
[31] Wu Z J, Zhao E J, Xiang H P, Hao X F, Liu X J, Meng J 2007 Phys. Rev. B 76 054115
Google Scholar
[32] Félix M, Coudert F X 2014 Phys. Rev. B 90 224104
Google Scholar
[33] Perdew J P, Wang Y 1992 Phys. Rev. B 45 13244
Google Scholar
[34] Heyd J, Scuseria G E, Ernzerhof M 2003 J. Chem. Phys. 118 8207
Google Scholar
[35] Smith N V 1971 Phys. Rev. B 3 1862
Google Scholar
[36] Draxl C A, Sofo J O 2006 Comput. Phys. Commun. 175 1
Google Scholar
[37] Ju M G, Dai J, Ma L, Zeng X C 2017 Adv. Energy Mater. 7 1700216
Google Scholar
[38] Zhang Z, Liu D W, Wu K C 2020 Spectrochim. Acta A. 226 117638
Google Scholar
[39] Mayengbama R, Tripathya S K, Palai G 2020 Mater. Today Commun. 24 101216
Google Scholar
[40] Liu Y, Qian J Y, Zhang H, Xu B, Zhang Y P, Liu L J, Chen G, Tian W J 2018 Org. Electron. 62 269
Google Scholar
-
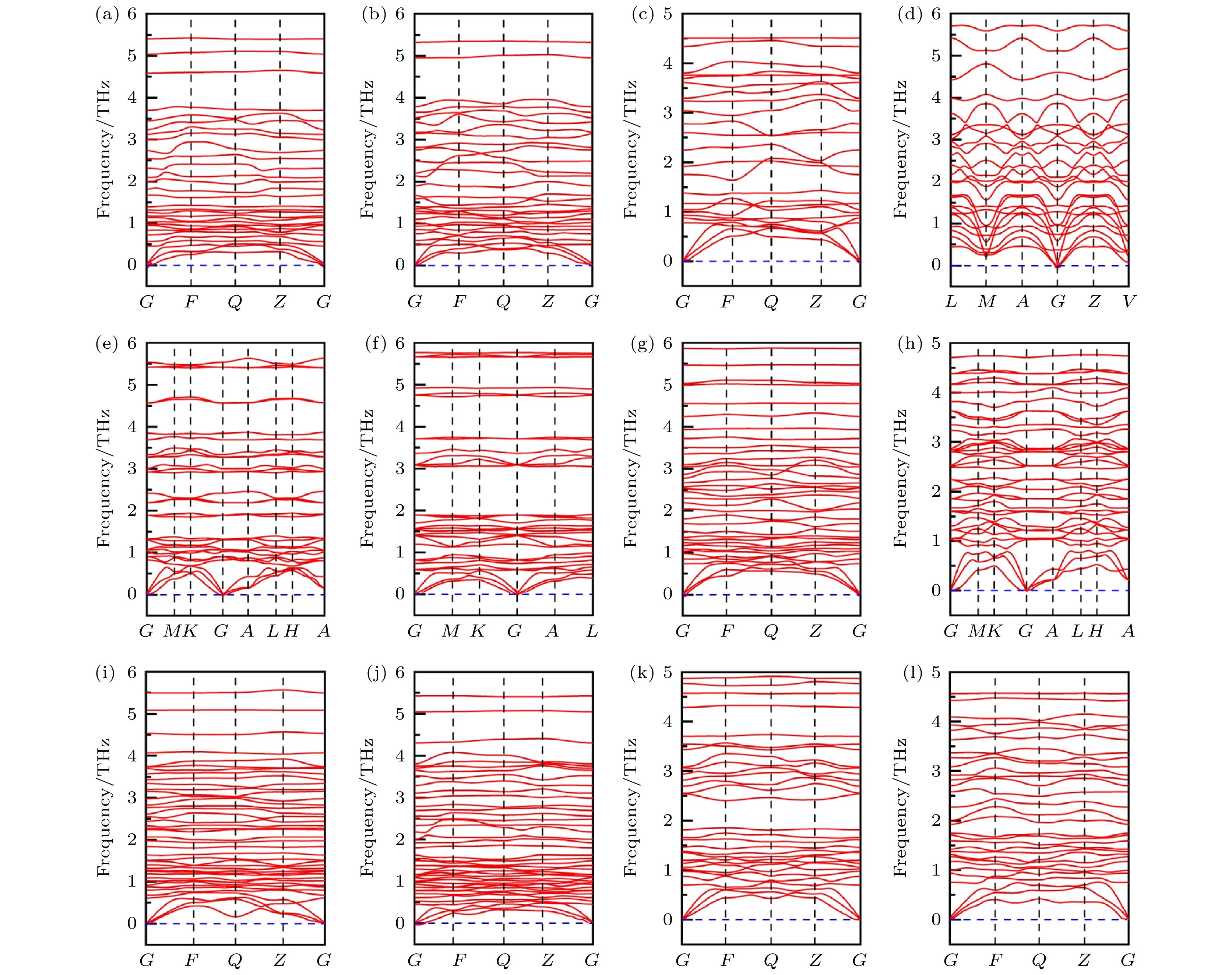
图 1 12个CuBiI三元化合物结构的声子色散谱图 (a) CuBi2I7-P1; (b) CuBi2I7-P1-II; (c) Cu2BiI5-P1; (d) Cu2BiI5-Cm; (e) Cu3BiI6-P3; (f) Cu3BiI6-R3; (g) Cu4BiI7-P1; (h) Cu4BiI7-P3; (i) Cu3Bi2I9-P1; (j) CuBi3I10-P1; (k) Cu2BiI7-P1; (l) Cu2BiI7-P1-II
Figure 1. Phonon dispersion spectra for the 12 structures of CuBiI ternary compound: (a) CuBi2I7-P1; (b) CuBi2I7-P1-II; (c) Cu2BiI5-P1; (d) Cu2BiI5-Cm; (e) Cu3BiI6-P3; (f) Cu3BiI6-R3; (g) Cu4BiI7-P1; (h) Cu4BiI7-P3; (i) Cu3Bi2I9-P1; (j) CuBi3I10-P1; (k) Cu2BiI7-P1; (l) Cu2BiI7-P1-II.
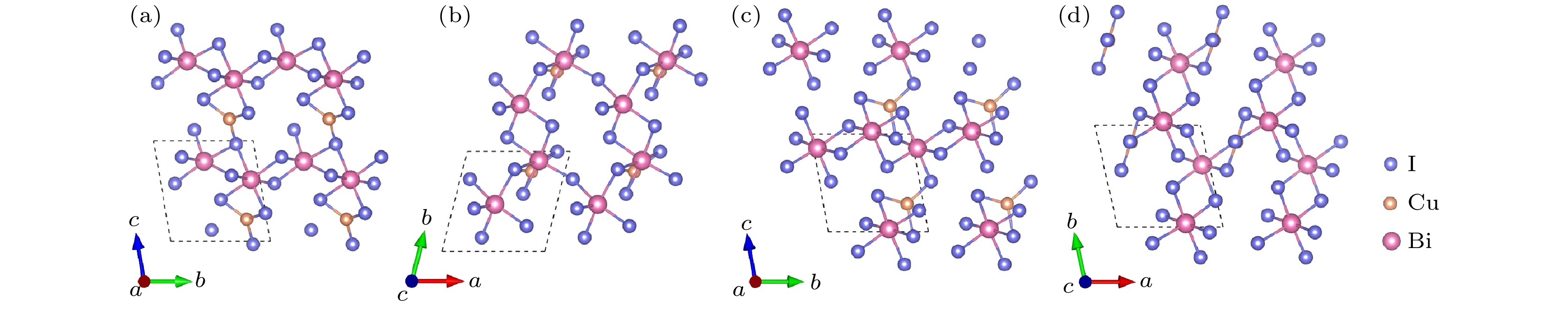
图 2 CuBi2I7-P1的晶体结构 (a) 主视图; (b) 俯视图. CuBi2I7-P1-II的晶体结构 (c) 主视图; (d) 俯视图
Figure 2. Crystal structure of CuBi2I7-P1: (a) Front view; (b) top view. Crystal structure of CuBi2I7-P1-II: (c) Front view; (d) top view

图 3 Cu2BiI5-P1的晶体结构 (a) 主视图; (b) 俯视图. Cu2BiI5-Cm的晶体结构 (c) 主视图; (d) 俯视图
Figure 3. Crystal structure of Cu2BiI5-P1: (a) Front view; (b) top view. Crystal structure of Cu2BiI5-Cm: (c) Front view; (d) top view.
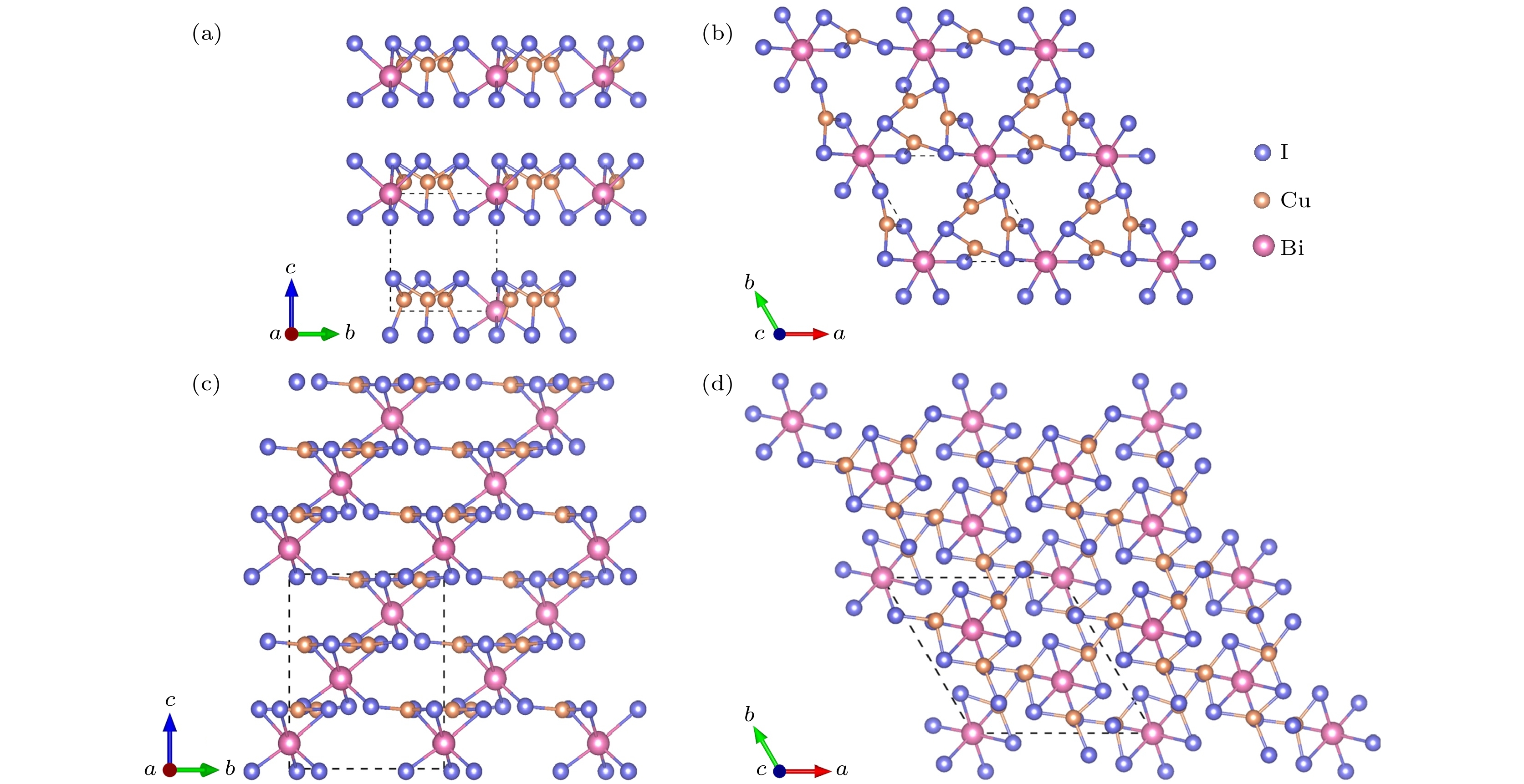
图 4 Cu3BiI6-P3的晶体结构 (a) 主视图; (b) 俯视图. Cu3BiI6-R3的晶体结构 (c) 主视图; (d) 俯视图
Figure 4. Crystal structure of Cu3BiI6-P3: (a) Front view; (b) top view. Crystal structure of Cu3BiI6-R3: (c) Front view; (d) top view.
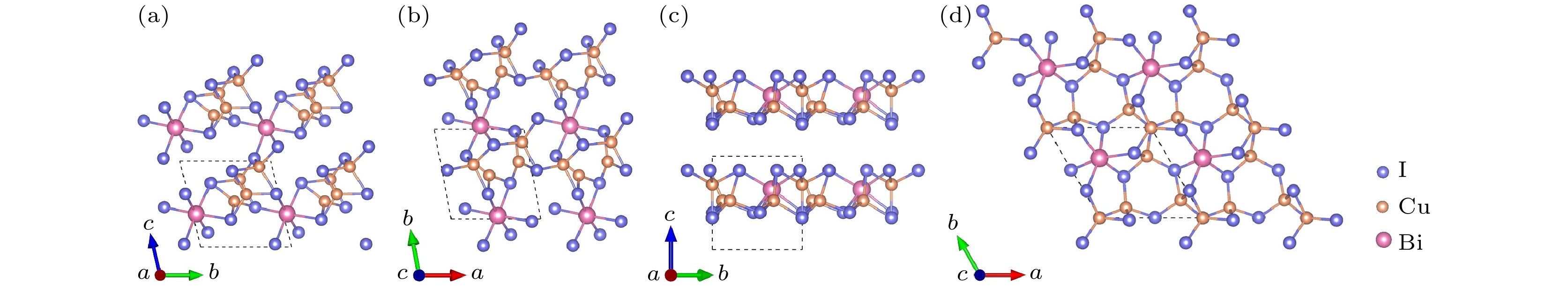
图 5 Cu4BiI7-P1的晶体结构 (a) 主视图; (b) 俯视图. Cu4BiI7-P3的晶体结构 (c) 主视图; (d) 俯视图
Figure 5. Crystal structure of Cu4BiI7-P1: (a) Front view; (b) top view. Crystal structure of Cu4BiI7-P3: (c) Front view; (d) top view.
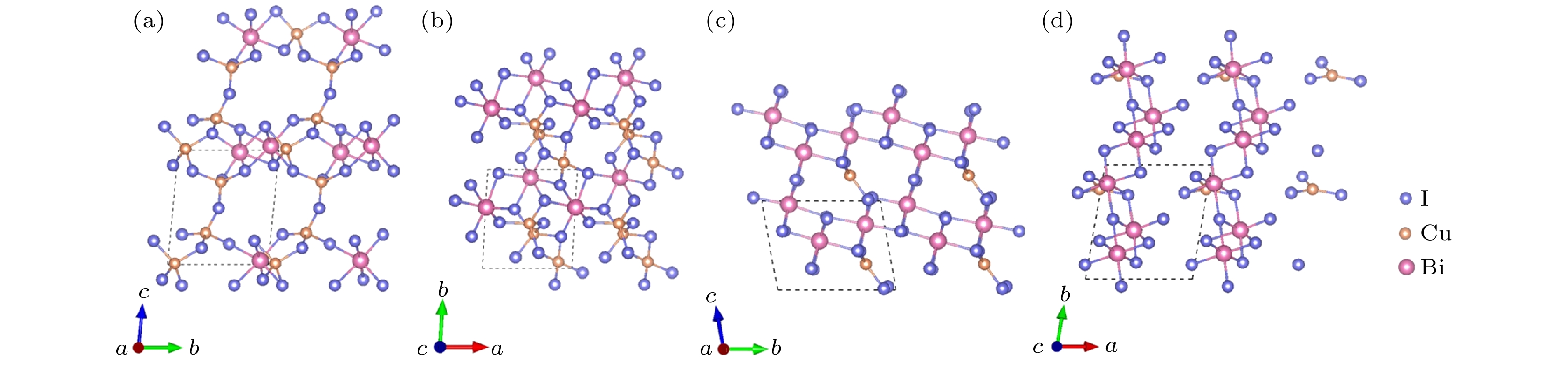
图 6 Cu3Bi2I9-P1的晶体结构 (a) 主视图; (b) 俯视图. CuBi3I10-P1的晶体结构 (c) 主视图; (d) 俯视图
Figure 6. Crystal structure of Cu3Bi2I9-P1: (a) Front view; (b) top view. Crystal structure of CuBi3I10-P1: (c) Front view; (d) top view
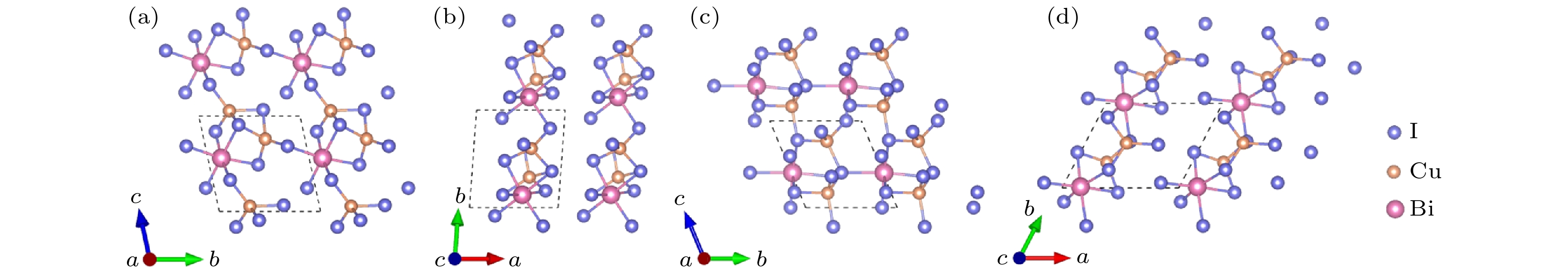
图 7 Cu2BiI7-P1的晶体结构 (a) 主视图; (b) 俯视图. Cu2BiI7-P1-II的晶体结构 (c) 主视图; (d) 俯视图
Figure 7. Crystal structure of Cu2BiI7-P1: (a) Front view; (b) top view. Crystal structure of Cu2BiI7-P1-II: (c) Front view; (d) top view
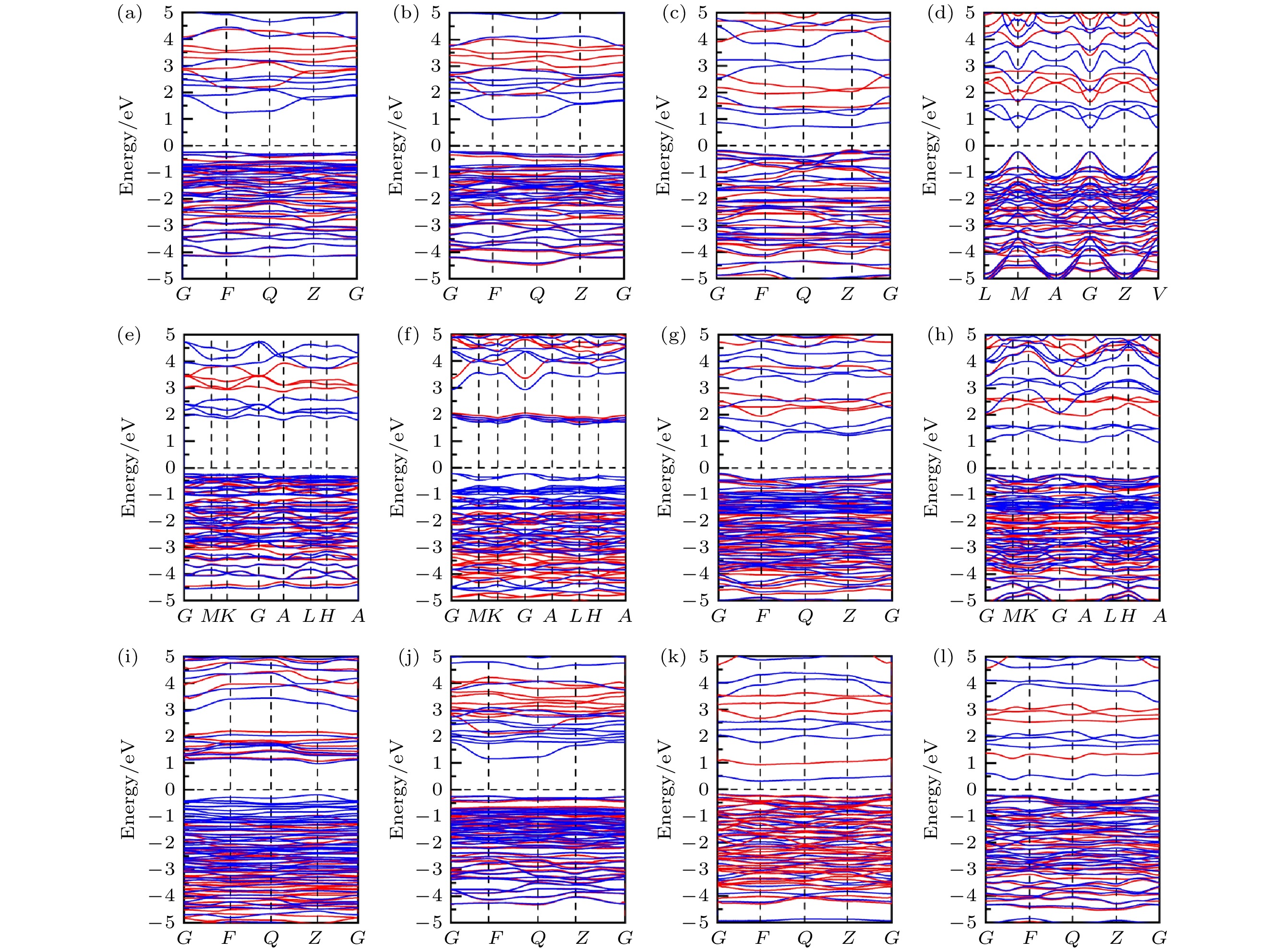
图 8 12个CuBiI三元化合物结构的能带结构图 (红色, HSE06方法计算结果; 蓝色, PBE方法计算结果) (a) CuBi2I7-P1; (b) CuBi2I7-P1-II; (c) Cu2BiI5-P1; (d) Cu2BiI5-Cm; (e) Cu3BiI6-P3; (f) Cu3BiI6-R3; (g) Cu4BiI7-P1; (h) Cu4BiI7-P3; (i) Cu3Bi2I9-P1; (j) CuBi3I10-P1; (k) Cu2BiI7-P1; (l) Cu2BiI7-P1-II
Figure 8. Band structure for the 12 structures of CuBiI ternary compound calculated by the PBE (blue lines) and HSE06 (red lines) methods: (a) CuBi2I7-P1; (b) CuBi2I7-P1-II; (c) Cu2BiI5-P1; (d) Cu2BiI5-Cm; (e) Cu3BiI6-P3; (f) Cu3BiI6-R3; (g) Cu4BiI7-P1; (h) Cu4BiI7-P3; (i) Cu3Bi2I9-P1; (j) CuBi3I10-P1; (k) Cu2BiI7-P1; (l) Cu2BiI7-P1-II.
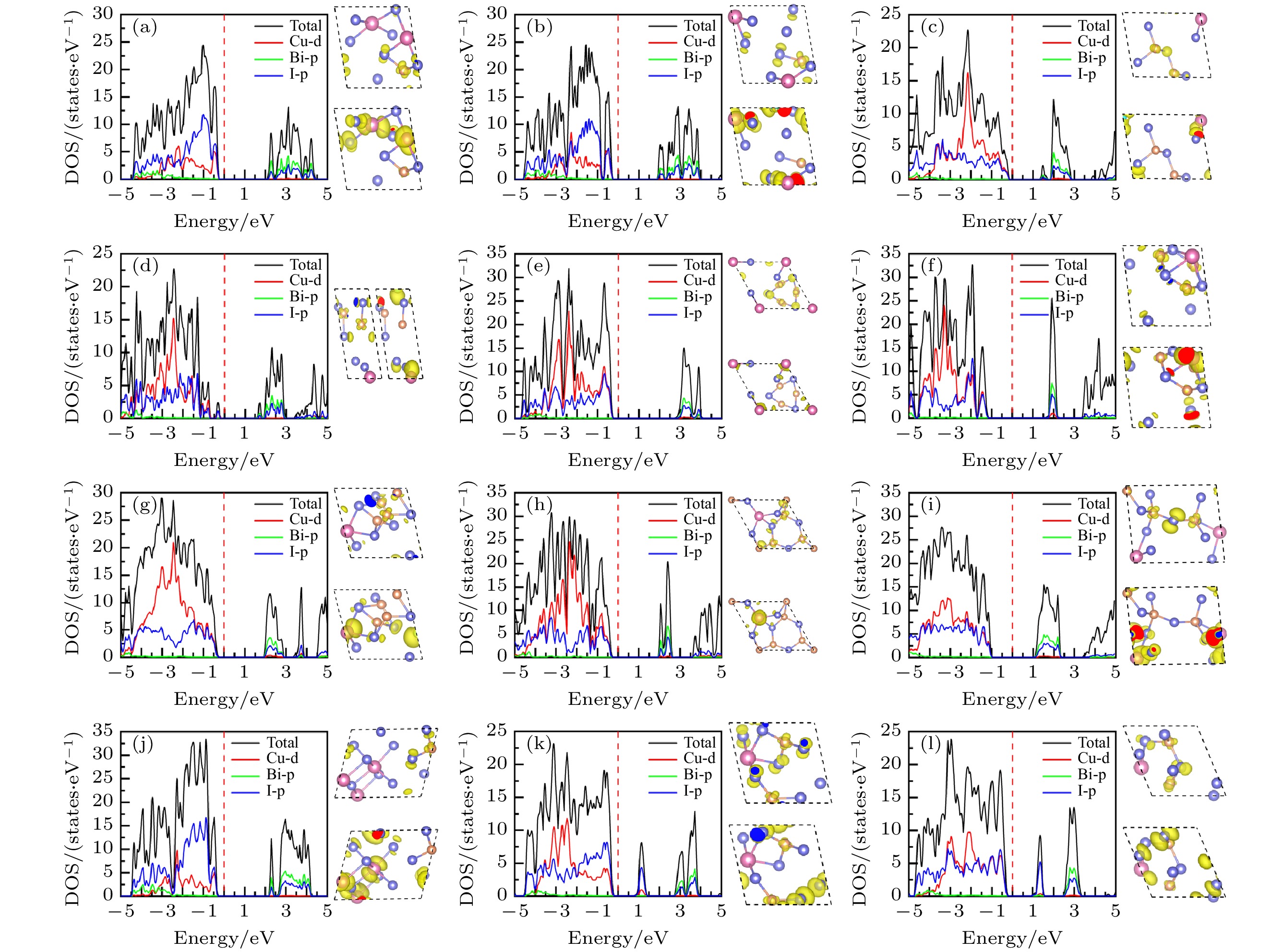
图 9 12个CuBiI三元化合物结构的总态密度、投影态密度图以及价带顶、导带底(从左到右或从上到下)的电荷密度分布图 (a) CuBi2I7-P1; (b) CuBi2I7-P1-II; (c) Cu2BiI5-P1; (d) Cu2BiI5-Cm; (e) Cu3BiI6-P3; (f) Cu3BiI6-R3; (g) Cu4BiI7-P1; (h) Cu4BiI7-P3; (i) Cu3Bi2I9-P1; (j) CuBi3I10-P1; (k) Cu2BiI7-P1; (l) Cu2BiI7-P1-II
Figure 9. Total density of state (TDOS), projection density of state (PDOS) and charge density distribution (Left to right or top to bottom) at CBM and VBM for the 12 structures of CuBiI ternary compound: (a) CuBi2I7-P1; (b) CuBi2I7-P1-II; (c) Cu2BiI5-P1; (d) Cu2BiI5-Cm; (e) Cu3BiI6-P3; (f) Cu3BiI6-R3; (g) Cu4BiI7-P1; (h) Cu4BiI7-P3; (i) Cu3Bi2I9-P1; (j) CuBi3I10-P1; (k) Cu2BiI7-P1; (l) Cu2BiI7-P1-II.
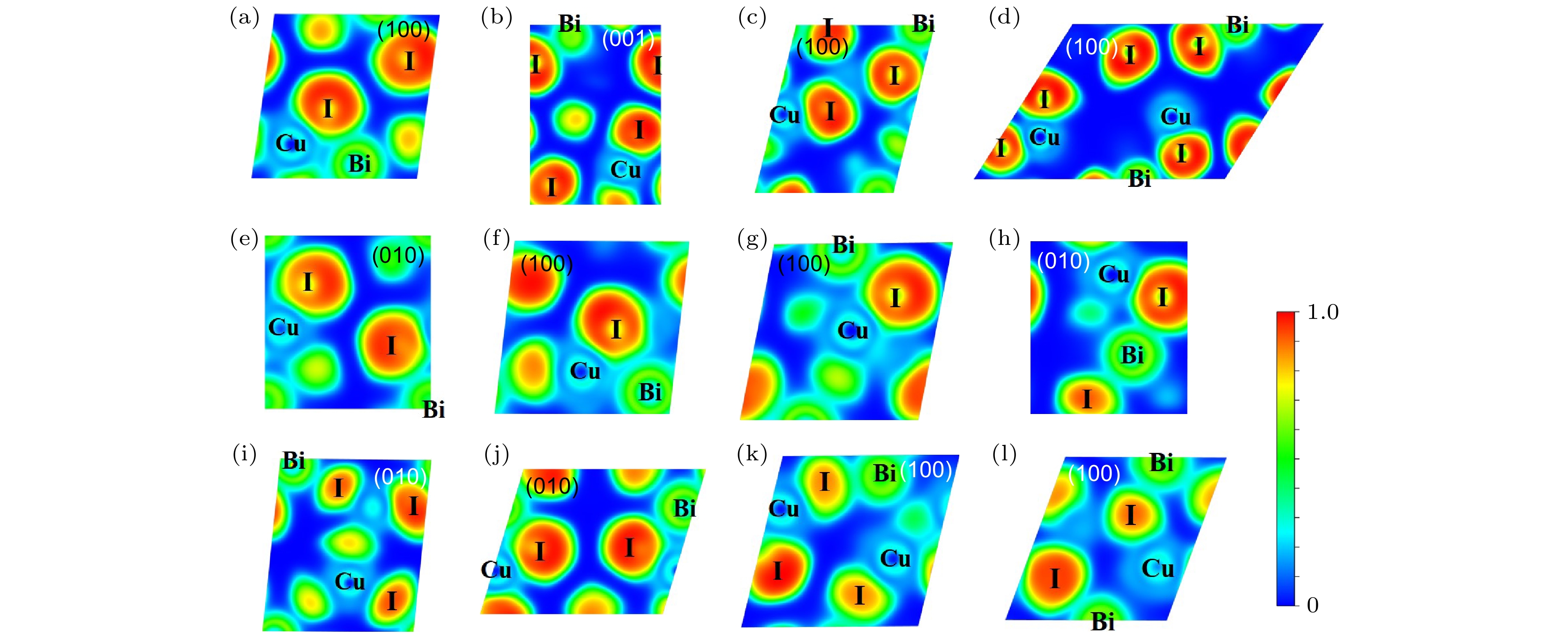
图 10 12个CuBiI三元化合物结构的电子局域函数分布图 (a) CuBi2I7-P1; (b) CuBi2I7-P1-II; (c) Cu2BiI5-P1; (d) Cu2BiI5-Cm; (e) Cu3BiI6-P3; (f) Cu3BiI6-R3; (g) Cu4BiI7-P1; (h) Cu4BiI7-P3; (i) Cu3Bi2I9-P1; (j) CuBi3I10-P1; (k) Cu2BiI7-P1; (l) Cu2BiI7-P1-II
Figure 10. Electron localization function (ELF) for the 12 structures of CuBiI ternary compound: (a) CuBi2I7-P1; (b) CuBi2I7-P1-II; (c) Cu2BiI5-P1; (d) Cu2BiI5-Cm; (e) Cu3BiI6-P3; (f) Cu3BiI6-R3; (g) Cu4BiI7-P3; (h) Cu4BiI7-P1; (i) Cu3Bi2I9-P1; (j) CuBi3I10-P1; (k) Cu2BiI7-P1; (l) Cu2BiI7-P1-II.
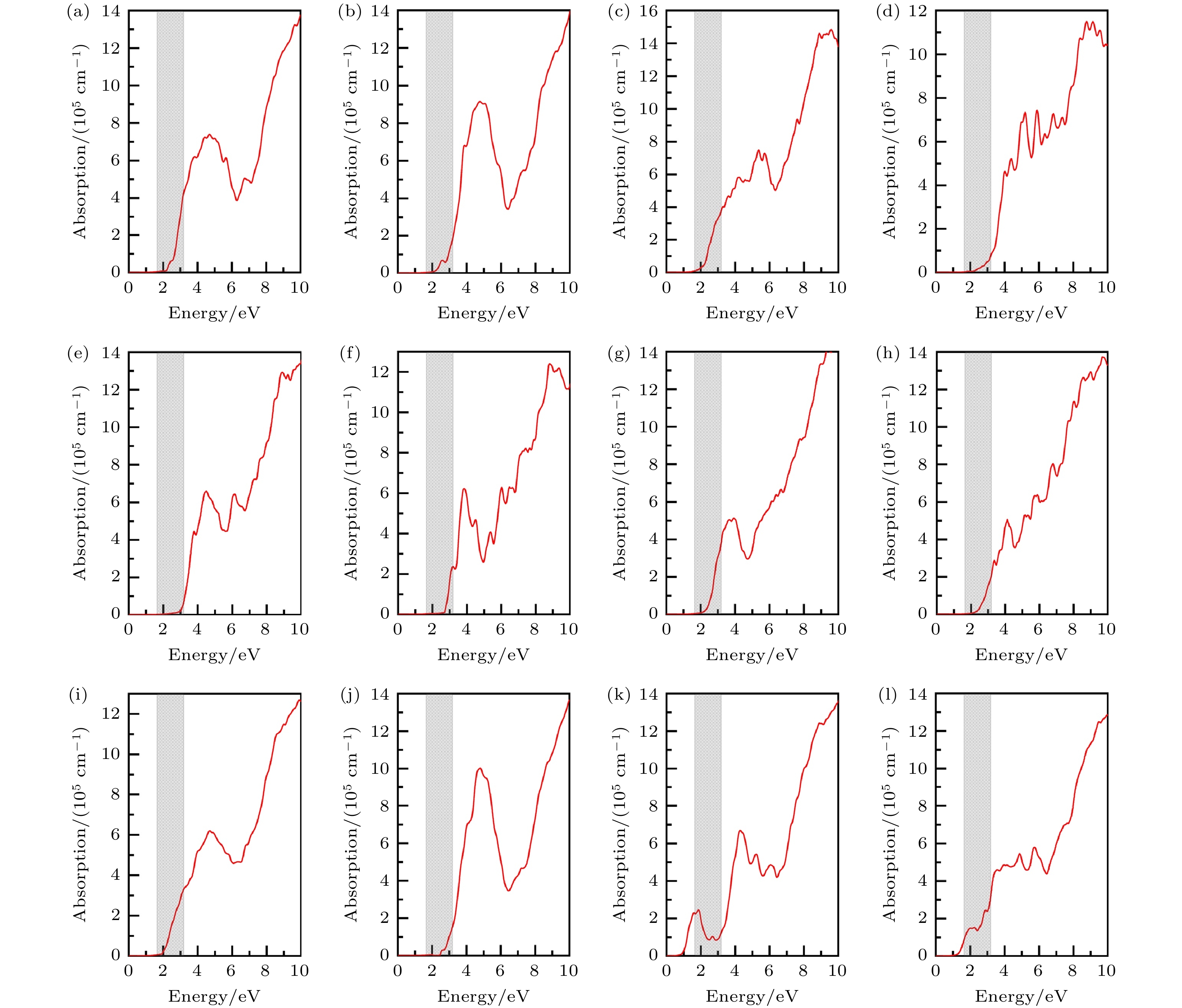
图 11 12个CuBiI三元化合物结构的光吸收谱, 灰色区域代表可见光能量范围(1.64—3.19 eV) (a) CuBi2I7-P1; (b) CuBi2I7-P1-II; (c) Cu2BiI5-P1; (d) Cu2BiI5-Cm; (e) Cu3BiI6-P3; (f) Cu3BiI6-R3; (g) Cu4BiI7-P1; (h) Cu4BiI7-P3; (i) Cu3Bi2I9-P1; (j) CuBi3I10-P1; (k) Cu2BiI7-P1; (l) Cu2BiI7-P1-II
Figure 11. Optical absorption spectrum for the 12 structures of CuBiI ternary compound. The gray area represents the Visible energy range (1.64–3.19 eV): (a) CuBi2I7-P1; (b) CuBi2I7-P1-II;(c) Cu2BiI5-P1; (d) Cu2BiI5-Cm; (e) Cu3BiI6-P3; (f) Cu3BiI6-R3; (g) Cu4BiI7-P1; (h) Cu4BiI7-P3; (i) Cu3Bi2I9-P1; (j) CuBi3I10-P1; (k) Cu2BiI7-P1; (l) Cu2BiI7-P1-II.
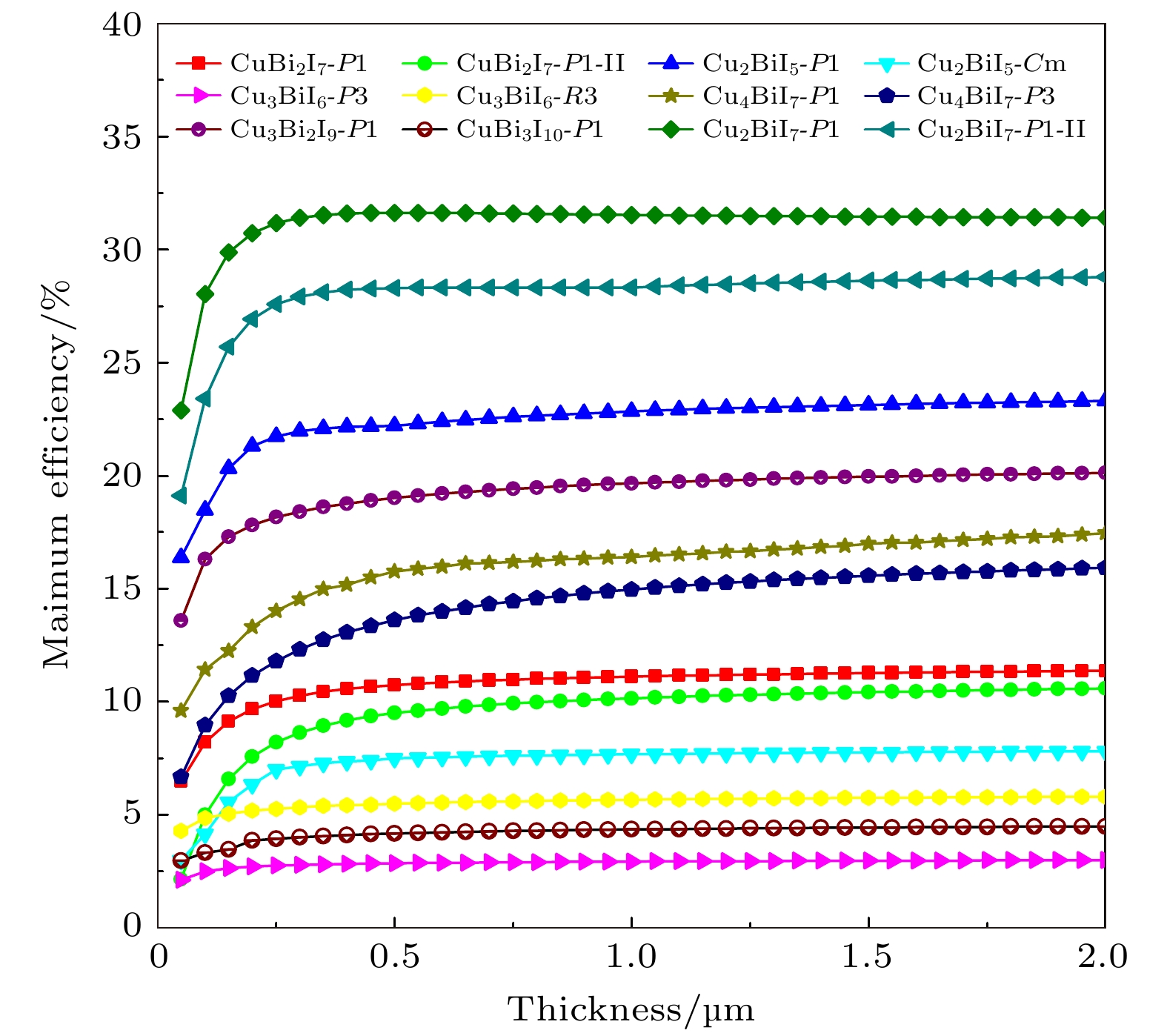
图 12 SLME方法预测的12个CuBiI三元化合物结构的光电转换效率与吸收层厚度的关系
Figure 12. Photoelectric conversion efficiency of 12 structures of CuBiI ternary compound with respect to the absorption layer thickness predicted by SLME method.
表 1 12个CuBiI三元化合物结构的结构名称、空间群、晶胞内原子数、体积及形成能
Table 1. Structure name, space group, number of atoms per unit cell, volume of the unit cell and formation energy for the 12 structures of CuBiI ternary compound.
Structure
nameSpace
groupNumber of/
(atoms·unit cell–1)Volume/
(Å3·unit cell–1)${{E} }_{\rm{form} }$/
(eV·atoms–1)Structure
nameSpace
groupNumber of/
(atoms·unit cell–1)Volume/
(Å3·unit cell–1)${{E} }_{\rm{form} }$/
(eV·atoms–1)CuBi2I7-P1 P1 10 474.24 –0.362 CuBi2I7-P1-II P1 10 465.35 –0.385 Cu2BiI5-P1 P1 8 295.03 –0.287 Cu2BiI5-Cm Cm 16 742.54 –0.290 Cu3BiI6-P3 P3 10 404.63 –0.265 Cu3BiI6-R3 R3 30 1318.62 –0.244 Cu4BiI7-P1 P1 12 428.29 –0.237 Cu4BiI7-P3 P3 12 451.33 –0.231 Cu3Bi2I9-P1 P1 14 645.41 –0.294 CuBi3I10-P1 P1 14 691.08 –0.402 Cu2BiI7-P1 P1 10 420.79 –0.225 Cu2BiI7-P1-II P1 10 420.68 –0.226  DownLoad: CSV
DownLoad: CSV
表 2 12个CuBiI三元化合物结构的弹性系数(Cij)
Table 2. Calculated elastic constants for the 12 structures of CuBiI ternary compound.
Cij/GPa CuBi2I7-P1 CuBi2I7-P1-II Cu2BiI5-P1 Cu2BiI5-Cm Cu3BiI6-P3 Cu3BiI6-R3 Cu4BiI7-P1 Cu4BiI7-P3 Cu3Bi2I9-P1 CuBi3I10-P1 Cu2BiI7-P1 Cu2BiI7-P1-II C11 8.34 9.03 39.90 4.76 11.99 17.59 23.63 32.25 17.16 2.82 9.71 3.12 C22 12.61 14.16 29.93 35.09 — — 18.34 — 20.42 9.34 14.12 10.34 C33 8.35 9.00 35.71 5.64 5.92 5.05 23.61 8.90 11.86 8.62 14.43 26.51 C44 3.52 3.62 10.18 1.73 1.01 3.53 6.87 1.41 3.91 3.27 6.46 7.16 C55 3.74 2.99 9.96 — — — 7.83 — 3.56 1.91 3.73 4.52 C66 2.41 4.43 6.77 1.24 4.42 6.13 8.72 11.26 6.01 1.93 3.13 3.18 C12 4.73 4.45 9.13 1.63 3.01 5.22 4.56 9.71 6.13 1.96 4.71 3.19 C13 2.61 2.57 14.13 2.77 1.33 2.99 7.76 3.30 3.14 2.11 5.61 6.45 C14 –2.07 –0.04 4.02 — 0.04 1.7 –1.84 0.28 0.51 –0.63 –0.17 –0.32 C15 0.18 0.21 0.18 –0.67 –0.15 –0.38 2.85 0.06 –0.79 –0.21 –2.11 0.54 C16 0.79 2.27 0.12 — — — –0.27 — –0.51 0.86 –1.73 –0.32 C23 2.69 2.76 14.94 2.42 — — 5.49 — 6.79 2.98 7.51 7.18 C24 –2.53 0.12 5.64 — — — –0.99 — 2.280 –0.05 –2.43 2.11 C25 0.28 0.16 0.17 –0.12 — — 2.68 — 0.49 –0.09 –3.12 1.54 C26 0.49 1.72 –0.02 — — — 0.04 — 0.41 1.47 1.35 1.01 C34 –1.69 –0.27 5.74 — — — –0.56 — 2.53 –0.89 –1.51 0.36 C35 –1.76 0.39 0.12 –0.90 — — 3.43 — –0.21 –2.38 –3.43 1.76 C36 1.47 1.17 –0.04 — — — 1.72 — 0.80 0.81 0.13 –0.48 C45 –0.41 0.83 0.13 — — — –0.47 — 0.21 0.85 1.26 0.19 C46 –0.10 0.42 0.01 0.12 — — 1.75 — 0.34 –0.62 –2.13 –0.69 C56 –0.58 –0.29 1.485 — — — –1.07 — 0.94 –0.53 0.82 0.72
DownLoad: CSV
表 3 12个CuBiI三元化合物结构的晶格常数以及Cu/Bi—I键长
Table 3. Lattice constants and Cu/Bi—I bond length for the 12 structures of CuBiI ternary compound.
Structure name a/Å b/Å c/Å α/(°) β/(°) γ/(°) Cu—I/Å Bi—I/Å CuBi2I7-P1 7.93 7.94 7.92 97.67 82.58 76.98 2.53—2.55 3.02—3.32 CuBi2I7-P1-II 8.05 7.85 7.75 97.64 100.86 100.78 2.54—2.55 3.03—3.22 Cu2BiI5-P1 4.42 7.62 9.57 95.94 103.35 106.82 2.59—2.67 3.09—3.18 Cu2BiI5-Cm 16.64 4.33 12.22 90.00 122.51 90.00 2.57—2.72 2.84—3.50 Cu3BiI6-P3 7.89 7.89 7.54 90.00 90.00 120.00 2.54—2.61 3.02—3.29 Cu3BiI6-R3 11.40 11.40 11.72 90.00 90.00 120.00 2.52—2.56 3.05—3.35 Cu4BiI7-P1 7.61 7.79 7.64 101.68 100.50 98.22 2.56—2.74 3.06—3.22 Cu4BiI7-P3 8.32 8.32 7.52 90.00 90.00 120.00 2.64—2.68 3.09—3.22 Cu3Bi2I9-P1 7.67 8.59 9.85 84.58 88.98 86.74 2.55—2.70 2.99—3.28 CuBi3I10-P1 9.46 10.12 7.85 103.09 106.70 77.25 2.53—2.54 2.99—3.32 Cu2BiI7-P1 7.33 7.90 7.92 104.09 108.49 81.74 2.59—2.64 3.05—3.34 Cu2BiI7-P1-II 9.00 7.78 7.20 109.91 89.24 64.61 2.58—2.70 2.98—3.34
DownLoad: CSV
表 4 12个CuBiI三元化合物结构的带隙值(HSE06和PBE方法计算结果), 价带顶与导带底位置, Bader电荷转移以及SLME (spectroscopic limited maximum efficiency)值
Table 4. Band gaps (Eg) calculated by the HSE06 and PBE method, positions of VBM and CBM, Bader charge and the spectroscopic limited maximum efficiency (SLME) values for the 12 structures of CuBiI ternary compound.
Structure name Eg/eV VBM CBM Bader charge SLME/% HSE06 PBE Cu/(e·atom–1) Bi/(e·atom–1) I/(e·atom–1) CuBi2I7-P1 2.39 1.48 0 0 0 0 0 0 0.33 1.08 –0.36 10.75 CuBi2I7-P1-II 2.13 1.21 0 0 0 0 0.5 0 0.33 1.09 –0.36 9.50 Cu2BiI5-P1 1.56 0.84 0 0 0.5 0 0.5 0 0.34 1.04 –0.34 22.20 Cu2BiI5-Cm 1.87 0.89 0 0 0 0 0 0 0.29 1.07 –0.33 7.50 Cu3BiI6-P3 3.09 1.97 0.05 0 0 0 0 0.5 0.29 1.08 –0.33 2.86 Cu3BiI6-R3 2.81 1.85 0 0 0 0.5 0 0.5 0.31 1.01 –0.32 5.49 Cu4BiI7-P1 2.19 1.22 0 0 0 0 0.5 0 0.30 1.03 –0.32 15.77 Cu4BiI7-P3 2.21 1.21 0 0 0.06 0 0 0.5 0.32 1.06 –0.33 13.61 Cu3Bi2I9-P1 2.03 1.17 0 0 0.5 0 0 0.5 0.34 1.02 –0.34 19.02 CuBi3I10-P1 2.36 1.41 0 0.5 0 0 0.5 0 0.33 1.09 –0.36 4.17 Cu2BiI7-P1 1.13 0.50 0 0 0 0 0.5 0 0.37 1.09 –0.26 31.63 Cu2BiI7-P1-II 1.40 0.60 0 0 0 0 0.5 0.5 0.35 1.06 –0.25 28.30
DownLoad: CSV
-
[1] Zhang F, Lu H P, Tong J H, Berry J J, Beard M C, Zhu K 2020 Energy Environ. Sci. 13 1154
Google Scholar
[2] Ajayan J, Nirmal D, Mohankumar P, Saravanan M, Jagadesh M, Arivazhagan L A 2020 Superlattices Microstruct. 143 106549
Google Scholar
[3] Wang L L, Fan B B, Zheng B, Yang Z B, Yin P G, Huo L J 2020 Sustainable Energy Fuels 4 2134
Google Scholar
[4] Chen W J, Li X Q, Li Y W, Li Y F 2020 Energy Environ. Sci. 13 1971
Google Scholar
[5] Wang Y, Sun H D 2018 Small Methods 21 700252
[6] Xi J, Wu Z X, Jiao B, Dong H, Ran C X, Piao C C, Lei T, Song T B, Ke W J, Yokoyama T, Hou X, Kanatzidis M G 2017 Adv. Mater. 29 1606964
Google Scholar
[7] Cheng P F, Wu T, Zhang J W, Li Y J, Liu J X, Jiang L, Mao X, Lu R F, Deng W Q, Han K L 2017 J. Phys. Chem. Lett. 8 4402
Google Scholar
[8] Sumathi R, Johnson K, Viswanathan B, Varadarajan T K 1998 Appl. Catal., A 172 15
Google Scholar
[9] Zhu H X, Liu J M 2016 Sci. Rep. 6 37425
Google Scholar
[10] Peng L, Xie W 2020 RSC Adv. 10 14679
Google Scholar
[11] Jiao Y Q, Lv Y Y, Li J, Niu M, Yang Z Q 2017 Comput. Theor. Chem. 15 20
[12] Zhang L, Liu C M, Lin Y 2019 J. Phys. Chem. Lett. 10 1676
Google Scholar
[13] Pa J, Bhunia A, Chakraborty S, Manna S, Das S, Dewan A, Datta S, Nag A 2018 J. Phys. Chem. C 122 10643
Google Scholar
[14] Sun S J, Tominaka S, Lee J H, Xie F, Bristowe P D, Cheetham A K 2016 APL Mater. 4 031101
Google Scholar
[15] Lu C J, Zhang J, Sun H R, Hou D G, Gan X L, Shang M H, Li Y Y, Hu Z Y, Zhu Y J, Han L Y 2018 ACS Appl. Energy Mater. 1 4485
Google Scholar
[16] Kulkarni A, Jena A K, Ikegami M 2019 Chem. Commun. 55 4031
Google Scholar
[17] Ramachandrana A A, Krishnana B D, Leal D A A, Martinez E G, Martinez J A A, Avellaneda D A, Shaji S 2020 Mater. Today Commun. 24 101092
Google Scholar
[18] Seo Y, Ha S R, Yoon S, Jeong S M, Choi H, Kang D W 2020 J. Power Sources 453 227903
Google Scholar
[19] Yi Z J, Zhang T, Ban H X, Shao H, Gong X, Wu M A, Liang G J, Zhang X L, Shen Y, Wang M K 2020 Sol. Energy 206 436
Google Scholar
[20] Fourcroy P H, Carré D, Thévet F, Rivet J 1991 Acta Crystallogr., Sect. C: Cryst. Struct. Commun. 47 2023
Google Scholar
[21] Hu Z S, Wang Z, Kapil G, Ma T L, Iikubo S, Minemoto T, Yoshino K, Toyoda T, Shen Q, Hayase S 2018 ChemSusChem 11 2930
Google Scholar
[22] Zhang B S, Lei Y, Qi R J, Yu H L, Yang X G, Cai T, Zheng Z 2019 Sci. China Mater. 62 519
Google Scholar
[23] Bi L Y, Hu Y Q, Li M Q, Hu T L, Zhang H L, Yin X T, Que W X, Lassoued M S, Zheng Y Z 2019 J. Mater. Chem. A. 7 19662
Google Scholar
[24] Lyakhov A O, Oganov A R, Stokes H T, Zhu Q 2013 Comput. Phys. Commun. 184 1172
Google Scholar
[25] Li Y L, Wang S N, Oganov A R, Gou H, Smith J S, Strobel T 2015 Nat. Commun. 6 6974
Google Scholar
[26] Kresse G, Furthmüller J 1996 Phys. Rev. B 54 11169
Google Scholar
[27] Perdew J P, Burke K, Ernzerhof M 1996 Phys. Rev. Lett. 77 3865
Google Scholar
[28] Blöchl P E 1994 Phys. Rev. B 50 17953
Google Scholar
[29] Monkhorst H J, Pack J D 1976 Phys. Rev. B 13 5188
Google Scholar
[30] Parlinski K, Li Z Q, Kawazoe Y 1997 Phys. Rev. Lett. 78 4063
Google Scholar
[31] Wu Z J, Zhao E J, Xiang H P, Hao X F, Liu X J, Meng J 2007 Phys. Rev. B 76 054115
Google Scholar
[32] Félix M, Coudert F X 2014 Phys. Rev. B 90 224104
Google Scholar
[33] Perdew J P, Wang Y 1992 Phys. Rev. B 45 13244
Google Scholar
[34] Heyd J, Scuseria G E, Ernzerhof M 2003 J. Chem. Phys. 118 8207
Google Scholar
[35] Smith N V 1971 Phys. Rev. B 3 1862
Google Scholar
[36] Draxl C A, Sofo J O 2006 Comput. Phys. Commun. 175 1
Google Scholar
[37] Ju M G, Dai J, Ma L, Zeng X C 2017 Adv. Energy Mater. 7 1700216
Google Scholar
[38] Zhang Z, Liu D W, Wu K C 2020 Spectrochim. Acta A. 226 117638
Google Scholar
[39] Mayengbama R, Tripathya S K, Palai G 2020 Mater. Today Commun. 24 101216
Google Scholar
[40] Liu Y, Qian J Y, Zhang H, Xu B, Zhang Y P, Liu L J, Chen G, Tian W J 2018 Org. Electron. 62 269
Google Scholar

 DownLoad:
DownLoad:
Catalog
Metrics
- Abstract views: 4988
- PDF Downloads: 142
- Cited By: 0
