










-
本文使用密度泛函理论中的广义梯度近似对扩展三明治结构graphene-2Li-graphene的几何结构、电子性质和储氢性能进行计算研究. 计算得知: 位于单层石墨烯中六元环面心位上方的单个Li原子与基底之间的结合能最大(1.19 eV), 但小于固体Li的实验内聚能(1.63 eV), 然而, 在双层石墨烯之间的单个Li原子与基底的结合能增加到3.41 eV, 远大于固体Li的实验内聚能, 因此位于双层石墨烯之间的多个Li原子不会成簇, 有利于进一步储氢. 扩展三明治结构graphene-2Li-graphene中每个Li原子最多可以吸附3个H2分子, 储氢密度高达10.20 wt.%, 超过美国能源部制定的5.5 wt.%的目标. 该体系对1—3 个H2分子的平均吸附能分别为0.37, 0.17和0.12 eV, 介于物理吸附和化学吸附(0.1—0.8 eV)之间, 因此该体系可以实现常温常压下对H2的可逆吸附. 通过对态密度分析可知, 每个Li原子主要通过电场极化作用吸附多个H2分子. 动力学和巨配分函数计算表明graphene-2Li-graphene结构对H2分子具有良好的可逆吸附性能. 该研究可以为开发良好的储氢材料提供一个好的研究思路, 为实验工作提供理论依据.The growth of population and the limited supply of fossil fuels have forced the world to seek for new kinds of alternative energy sources which are abundant, renewable, efficient, secure and pollution-free. In this regard, hydrogen is generally considered as a potential candidate. However, it is a great challenge to find hydrogen storage materials with large hydrogen gravimetric density under ambient thermodynamic conditions. The most effective way to improve the hydrogen storage capacity is to decorate the pure nanomaterials with transition metals, alkaline metals, and alkaline earth metals. The generalized gradient approximation based on density functional theory is used to study the hydrogen storage capacity of the expanded sandwich structure graphene-2Li-graphene. It is calculated that the structure with the Li atom located above the face site of the hexagonal ring of the graphene has the maximum binding energy (1.19 eV), which is less than the experimental cohesive energy of bulk Li (1.63 eV). However, the calculated binding energy values of the Li atom to the upper and lower graphene layer are both 3.43 eV, which is much larger than the experimental cohesive energy value of bulk Li, so it can prevent the Li atoms from clustering between graphene layers. Each Li atom in the graphene-2Li-graphene structure can adsorb 3 H2 molecules at most. Thus, the hydrogen gravimetric density of graphene-2(Li-3H2)-graphene is 10.20 wt.%, which had far exceeded the gravimetric density of the target value of 5.5 wt.% by the year 2017 specified by the US Department of Energy. The average adsorption energy values of H2 adsorbed per Li are 0.37, 0.17, and 0.12 eV respectively for 1−3 H2 molecules, which are between the physical adsorption and chemical adsorption(0.1−0.8 eV), therefore, it can realize the reversible adsorption of hydrogen. Each Li atom can adsorb 3 H2 molecules at most by the electronic polarization interaction. The dynamic calculations and GFRF calculations show that the interlayer Li atom doped double-layer graphene has good reversible adsorption performance for hydrogen. This research can provide a good research idea for developing good hydrogen storage materials and theoretical basis for experimental worker. These findings can suggest a way to design hydrogen storage materials under the near-ambient conditions.
-
Keywords:
- graphene /
- Li /
- electronic properties /
- hydrogen storage /
- density functional theory
[1] Schlapbach L, Zuttel A 2001 Nature 414 353
 Google Scholar
Google Scholar
[2] Chandrakumar K R S, Ghosh S K 2008 Nano Lett. 8 13
Google Scholar
[3] Rosi N L, Eckert J, Eddaoudi M, Vodak D T, Kim J, O'keeffe M, Yaghi O M 2003 Science 300 1127
Google Scholar
[4] Han S S, Goddard W A 2007 J. Am. Chem. Soc. 129 8422
Google Scholar
[5] Kealy T J, Pauson P L 1951 Nature 168 1039
[6] Sun Q, Wang Q, Jena P, Kawazoe Y 2005 J. Am. Chem. Soc. 127 14582
Google Scholar
[7] Kim D, Lee S, Hwang Y, Yun K H, Chung Y C 2014 Int. J. Hydrogen Energy 39 13189
Google Scholar
[8] Xu B, Lei X L, Liu G, Wu M S, Ouyang C Y 2014 Int. J. Hydrogen Energy 39 17104
Google Scholar
[9] Seenithurai S, Pandyan R K, Kumar S V, Saranya C, Mahendran M 2014 Int. J. Hydrogen Energy 39 11016
Google Scholar
[10] Chen L, Zhang Y, Koratkar N, Jena P, Nayak S K 2008 Phys. Rev. B 77 033405
[11] Mauron P, Gaboardi M, Remhof A, Bliersbach A, Sheptyakov D, Aramini M, Vlahopoulou G, Giglio F, Pontiroli D, Ricco M, Zuttel A 2013 J. Phys. Chem. C 117 22598
Google Scholar
[12] Lein M, Frunzke J, Frenking G 2003 Inorg. Chem. 42 2504
Google Scholar
[13] Youn I S, Kim D Y, Singh N J, Park S W, Youn J, Kim K S 2011 J. Chem. Theor. Comput. 8 99
[14] Kealy T J, Pauson P L 1951 Nature 168 1039
[15] Wilkinson G, Rosenblum M, Whiting M C, Woodward R B 1952 J. Am. Chem. Soc. 74 2125
Google Scholar
[16] Kubas G J 2001 Kluwer Academic (New York: Plenum Publishing)
[17] Lein M, Frunzke J, Frenking G 2003 Inorg. Chem. 42 2504
Google Scholar
[18] Youn I S, Kim D Y, Singh N J, Park S W, Youn J, Kim K S 2011 J. Chem. Theory Comput. 8 99
[19] Sun Q, Wang Q, Jena P, Kawazoe Y 2005 J. Am. Chem. Soc. 127 14582
Google Scholar
[20] Delley B 1990 J. Chem. Phys. 92 508
Google Scholar
[21] Zhang Q Y, Tang C M, Zhu W H, Cheng C 2018 J. Phys. Chem. C 122 22838
Google Scholar
[22] Chang L T, Wei C, Xiao H T 2006 Chin. Phys. 15 2718
Google Scholar
[23] Zhao J Y, Zhao F Q, Xu S Y, Ju X H 2013 J. Phys. Chem. A 117 2213
Google Scholar
[24] Grimme S, Antony J, Ehrlich S, Krieg H 2010 J. Chem. Phys. 132 154104
Google Scholar
[25] Ma L, Zhang J M, Xu K W 2014 Appl. Surf. Sci. 292 921
Google Scholar
[26] Gao Y, Wu X, Zeng X C 2014 J. Mater. Chem. A 2 5910
Google Scholar
[27] Park J, Burova S, Rodgers A S, Lin M C 1999 J. Phys. Chem. A 103 9036
Google Scholar
[28] Abad E, Dappe Y J, Martínez J I, Flores F, Ortega J 2011 J. Chem. Phys. 134 044701
Google Scholar
[29] Pliva J, Johns J W C, Goodman L 1991 J. Mol. Spectrosc. 148 427
Google Scholar
[30] Toyoda K, Nakano Y, Hamada I, Lee K, Yanagisawa S, Morikawa Y 2009 Surf. Sci. 603 2912
Google Scholar
[31] Wang X B, Tang C M, Zhu W H, Zhou X F, Zhou Q H, Cheng C 2018 J. Phys. Chem. C 122 9654
Google Scholar
[32] Kealy T J, Pauson P L 1951 Nature 168 1039
[33] Wu G, Wang J, Zhang X, Zhu L 2009 J. Phys. Chem. C 113 7052
Google Scholar
[34] 汪志诚 2013 热力学·统计物理 (第五版) (北京: 高等教育出版社)
Wang Z C 2013 Thermodynamics·Statistical Physics (5th Ed.) (Beijing: Higher Education Press) (in Chinese)
-
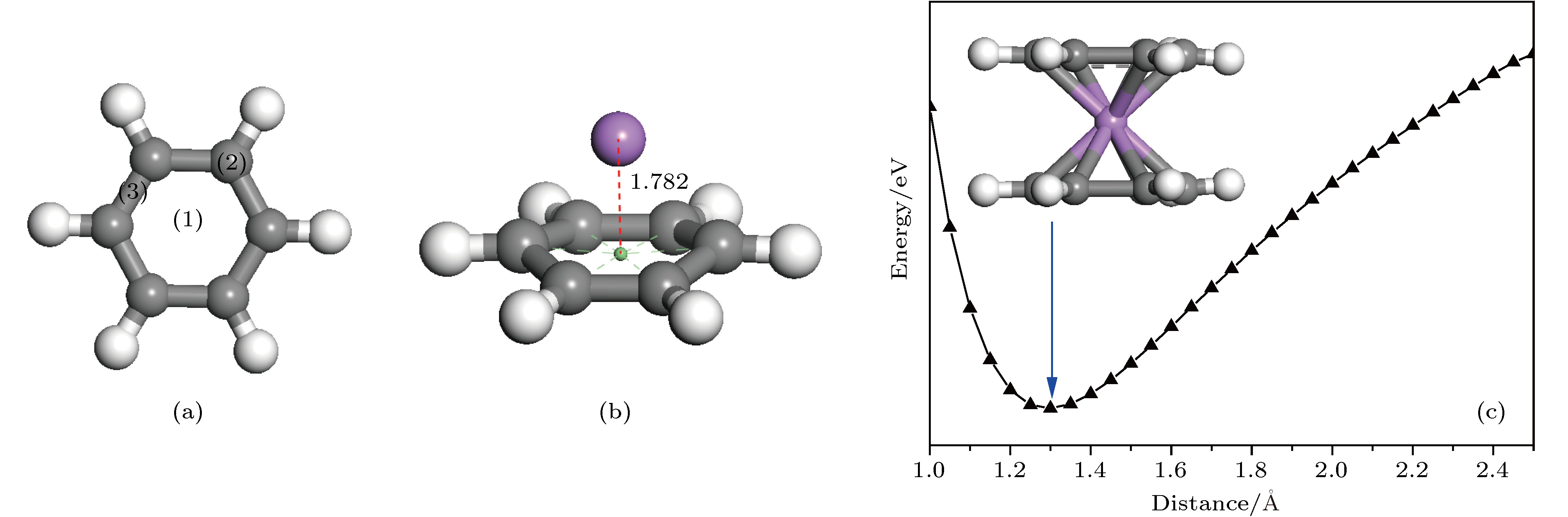
图 1 (a)苯环中3种不等价位置; (b) Li原子位于苯环面心位上方的优化结构; (c) C6H6-Li-C6H6势能面扫描曲线和最稳定的C6H6-Li-C6H6三明治结构
Fig. 1. (a) Three unequal positions in benzene ring; (b) the optimized structure of the benzene ring with the Li atom located above the face site of the hexagonal ring; (c) potential energy surface scanning curve of C6H6-Li-C6H6 and the most stable sandwich structure of C6H6-Li-C6H6.
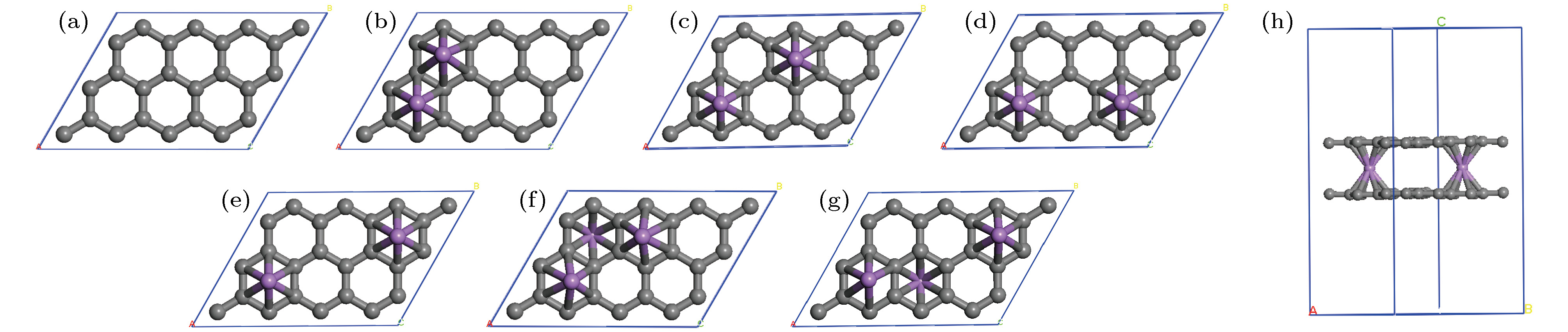
图 2 (a) 单层石墨烯的2 × 3晶胞中Li原子的6个位置; 2个Li原子分别位于(b) ①④组合; (c) ①⑤组合; (d) ①③组合; (e) ①⑥组合; 3个Li原子分别位于(f) ①④⑤组合; (g) ①②⑥组合; (h) 2个Li原子掺杂的最稳定双层石墨烯结构graphene-2Li-graphene
Fig. 2. (a) 6 positions of the Li atom in the 2 × 3 unit cell of monolayer graphene; two Li atoms are located at (b) ①④ combination; (c) ①⑤ combination, (d) ①③ combination; (e) ①⑥ composition; three Li atoms are located in (f) ①④⑤ combination; (g) ①②⑥ combination; (h) the most stable graphene-2Li-graphene double-layer graphene structure doped by two Li atoms.

图 3 graphene-2Li-graphene 的2 × 3晶胞中每个Li原子分别吸附1—4个H2分子的结构图
Fig. 3. The structural of the 2 × 3 unit cell of graphene-2Li-graphene with each Li atom adsorbed by 1−4 H2 molecules.
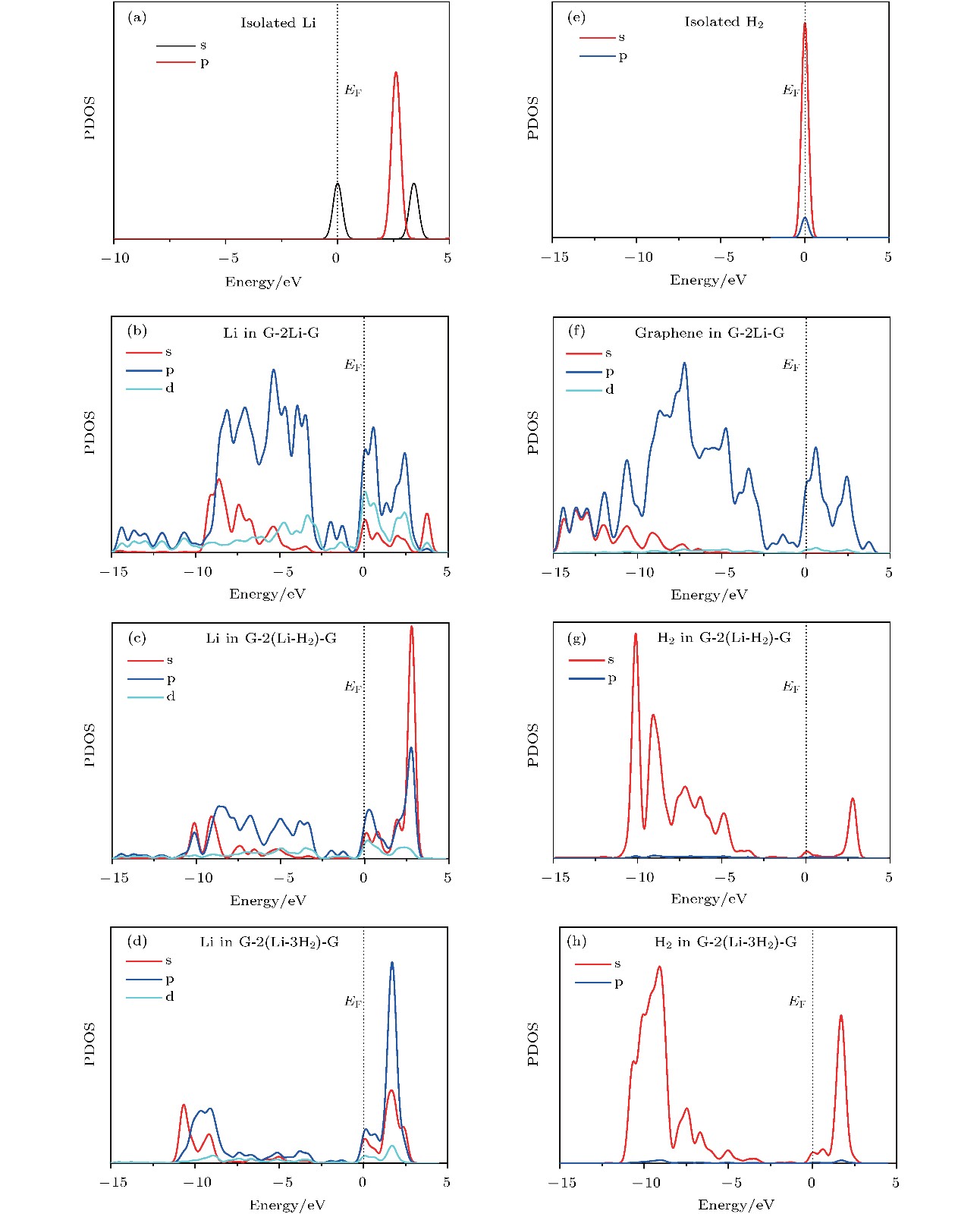
图 4 不同结构中Li原子或H2分子的态密度图 (a)单独的Li原子; (b) graphene-2Li-graphene中Li原子; (c) graphene-2(Li-H2)-graphene中Li原子; (d) graphene-2(Li-3H2)-graphene中Li原子; (e) 单独的H2分子; (f) graphene-2Li-graphene中graphene; (g) graphene-2(Li-H2)-graphene中H2分子; (h) graphene-2(Li-3H2)-graphene中H2分子
Fig. 4. The PDOS of Li atom or H2 molecules: (a) Isolated Li atom; (b) the Li atom in graphene-2Li-graphene; (c) the Li atom in graphene-2(Li-H2)-graphene; (d) the Li atom in graphene-2(Li-3H2)-graphene; (e) isolated H2 molecules; (f) the graphene in graphene-2Li-graphene; (g) the H2 molecules in graphene-2(Li-H2)-graphene; (h) the H2 molecules in graphene-2(Li-3H2)-graphene.
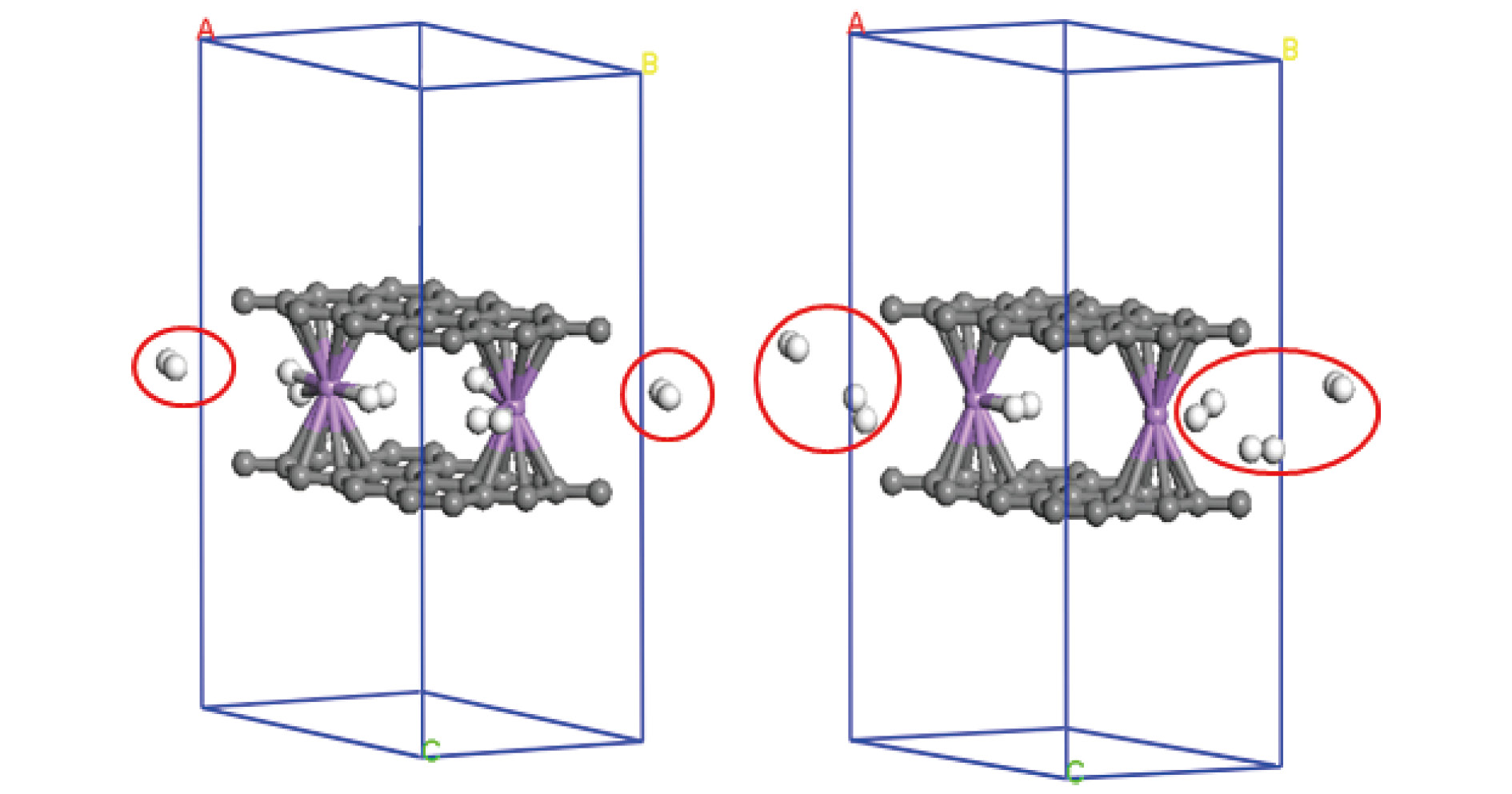
图 5 graphene-2(Li-3H2)-graphene在77和300 K下5 ps之后的动力学结构图
Fig. 5. The structures of graphene-2(Li-3H2)-graphene at 77 and 300 K after 5 ps dynamic times.
表 1 扩展三明治结构graphene-2(Li-nH2)-graphene[G-2(Li-nH2)-G)](n = 1—4)中的H2分子的Ead, Er, Li和H的平均bader电荷(QLi和QH), 双层石墨烯的层间距(DG-G)
Table 1. The Ead and Er of H2 molecules average bader charge of Li and H (QLi and QH), interlayer distance of double-layer graphene (DG-G) in the expanded sandwich structure graphene-2(Li-nH2)-graphene[G-2(Li-nH2)-G)](n = 1—4).
G-2Li-G G-2(Li-H2)-G G-2(Li-2H2)-G G-2(Li-3H2)-G G-2(Li-4H2)-G Ead/eV — 0.37 0.17 0.12 0.06 Er/eV — 0.19 0.19 0.10 –0.08 QLi/e 0.99 0.62 0.31 0.02 0.01 QH/e — 0.20 0.18 0.16 0.12 DG-G/Å 3.69 3.84 4.40 4.90 4.93  下载: 导出CSV
下载: 导出CSV
-
[1] Schlapbach L, Zuttel A 2001 Nature 414 353
Google Scholar
[2] Chandrakumar K R S, Ghosh S K 2008 Nano Lett. 8 13
Google Scholar
[3] Rosi N L, Eckert J, Eddaoudi M, Vodak D T, Kim J, O'keeffe M, Yaghi O M 2003 Science 300 1127
Google Scholar
[4] Han S S, Goddard W A 2007 J. Am. Chem. Soc. 129 8422
Google Scholar
[5] Kealy T J, Pauson P L 1951 Nature 168 1039
[6] Sun Q, Wang Q, Jena P, Kawazoe Y 2005 J. Am. Chem. Soc. 127 14582
Google Scholar
[7] Kim D, Lee S, Hwang Y, Yun K H, Chung Y C 2014 Int. J. Hydrogen Energy 39 13189
Google Scholar
[8] Xu B, Lei X L, Liu G, Wu M S, Ouyang C Y 2014 Int. J. Hydrogen Energy 39 17104
Google Scholar
[9] Seenithurai S, Pandyan R K, Kumar S V, Saranya C, Mahendran M 2014 Int. J. Hydrogen Energy 39 11016
Google Scholar
[10] Chen L, Zhang Y, Koratkar N, Jena P, Nayak S K 2008 Phys. Rev. B 77 033405
[11] Mauron P, Gaboardi M, Remhof A, Bliersbach A, Sheptyakov D, Aramini M, Vlahopoulou G, Giglio F, Pontiroli D, Ricco M, Zuttel A 2013 J. Phys. Chem. C 117 22598
Google Scholar
[12] Lein M, Frunzke J, Frenking G 2003 Inorg. Chem. 42 2504
Google Scholar
[13] Youn I S, Kim D Y, Singh N J, Park S W, Youn J, Kim K S 2011 J. Chem. Theor. Comput. 8 99
[14] Kealy T J, Pauson P L 1951 Nature 168 1039
[15] Wilkinson G, Rosenblum M, Whiting M C, Woodward R B 1952 J. Am. Chem. Soc. 74 2125
Google Scholar
[16] Kubas G J 2001 Kluwer Academic (New York: Plenum Publishing)
[17] Lein M, Frunzke J, Frenking G 2003 Inorg. Chem. 42 2504
Google Scholar
[18] Youn I S, Kim D Y, Singh N J, Park S W, Youn J, Kim K S 2011 J. Chem. Theory Comput. 8 99
[19] Sun Q, Wang Q, Jena P, Kawazoe Y 2005 J. Am. Chem. Soc. 127 14582
Google Scholar
[20] Delley B 1990 J. Chem. Phys. 92 508
Google Scholar
[21] Zhang Q Y, Tang C M, Zhu W H, Cheng C 2018 J. Phys. Chem. C 122 22838
Google Scholar
[22] Chang L T, Wei C, Xiao H T 2006 Chin. Phys. 15 2718
Google Scholar
[23] Zhao J Y, Zhao F Q, Xu S Y, Ju X H 2013 J. Phys. Chem. A 117 2213
Google Scholar
[24] Grimme S, Antony J, Ehrlich S, Krieg H 2010 J. Chem. Phys. 132 154104
Google Scholar
[25] Ma L, Zhang J M, Xu K W 2014 Appl. Surf. Sci. 292 921
Google Scholar
[26] Gao Y, Wu X, Zeng X C 2014 J. Mater. Chem. A 2 5910
Google Scholar
[27] Park J, Burova S, Rodgers A S, Lin M C 1999 J. Phys. Chem. A 103 9036
Google Scholar
[28] Abad E, Dappe Y J, Martínez J I, Flores F, Ortega J 2011 J. Chem. Phys. 134 044701
Google Scholar
[29] Pliva J, Johns J W C, Goodman L 1991 J. Mol. Spectrosc. 148 427
Google Scholar
[30] Toyoda K, Nakano Y, Hamada I, Lee K, Yanagisawa S, Morikawa Y 2009 Surf. Sci. 603 2912
Google Scholar
[31] Wang X B, Tang C M, Zhu W H, Zhou X F, Zhou Q H, Cheng C 2018 J. Phys. Chem. C 122 9654
Google Scholar
[32] Kealy T J, Pauson P L 1951 Nature 168 1039
[33] Wu G, Wang J, Zhang X, Zhu L 2009 J. Phys. Chem. C 113 7052
Google Scholar
[34] 汪志诚 2013 热力学·统计物理 (第五版) (北京: 高等教育出版社)
Wang Z C 2013 Thermodynamics·Statistical Physics (5th Ed.) (Beijing: Higher Education Press) (in Chinese)

 下载:
下载:
计量
- 文章访问数: 15273
- PDF下载量: 108
- 被引次数: 0
