










-
基于第一性原理深入研究了碱金属原子(Li, Na, K)修饰的多孔石墨烯(PG)体系的储氢性能, 并且通过从头算分子动力学模拟了温度对Li-PG吸附的H2分子稳定性的影响. 研究结果表明, PG结构的碳环中心是碱金属原子最稳定的吸附位置, PG单胞最多可以吸附4个碱金属原子, Li原子被束缚最强, 金属原子间无团聚的倾向; H2分子通过极化机制吸附在碱金属修饰的PG结构上, 每个金属原子周围最多可以稳定地吸附3个H2分子; Li-PG对H2分子的吸附最强(平均吸附能为–0.246 eV/H2), Na-PG对H2分子的吸附较弱(平均吸附能为–0.129 eV/H2), K-PG对H2分子的吸附最弱(平均吸附能为–0.056 eV/H2), 不适合用做储氢材料; 在不考虑外界压强且温度为300 K的情况下, Li-PG结构可稳定地吸附9个H2分子, 储氢量为9.25 wt.%; 在400 K时, 有7个吸附H2分子脱离Li-PG的束缚, 在600—700 K的范围内, 吸附H2分子全部脱离了Li-PG体系的束缚.Porous graphene (PG), a kind of graphene-related material with nanopores in the graphene plane, exhibits novel properties different from those of pristine graphene, leading to its potential applications in many fields. Owing to periodic nanopores existing naturally in the two-dimensional layer, PG can be used as an ideal candidate for hydrogen storage material. High hydrogen storage capacity of Li-decorated PG has been investigated theoretically, but the effect of temperature on the stability of the H2 adsorbed on Li-PG has been not discussed yet. In this paper, by using the first-principles method, the hydrogen storage capacity on alkaline metal atoms (Li, Na, K) decorated porous graphene is investigated in depth with generalized gradient approximation, and the effect of the temperature on the stability of the hydrogen adsorption system is elucidated by the ab initio molecular-dynamics simulation. The results show that the most favorable adsorption sites of Li, Na and K are the hollow center sites of the C hexagon, and four alkaline metal atoms can be adsorbed stably on both sides of PG unit cell without clustering. Alkaline metal adatoms adsorbed on PG become positively charged by transferring charge to PG and adsorbed H2 molecules, and three H2 molecules can be adsorbed around each alkaline metal atom. By analyzing the Mulliken atomic populations, charge density differences and density of states of H2 adsorbed on Li-PG system, we find that the H2 molecules are adsorbed on alkaline metal atoms decorated graphene complex by attractive interaction between positively charged alkaline metal adatoms and negatively charged H and weak van der Waals interaction. Twelve H2 molecules are adsorbed on both sides of PG decorated with alkaline metal atoms. The average adsorption energy of H2 adsorbed on Li-PG, Na-PG and K-PG are –0.246, –0.129 and –0.056 eV/H2, respectively. It is obvious that the hydrogen adsorption capacity of Li-PG system is strongest, and the hydrogen adsorption capacity of K-PG is weakest, thus K-PG structure is not suitable for hydrogen storage. Furthermore, by the ab initio molecular-dynamic simulation, in which the NVT ensemble is selected but the external pressure is not adopted, the effect of temperature on the stability of H2 molecules adsorbed on Li-PG system is elucidated. The result shows that the configuration of Li-PG is very stable, H2 molecules are stably adsorbed around the Li atoms at low temperature, and some H2 molecules start to be desorbed from the Li atoms with the increase of temperature. At 200 K, H2 molecules begin to move away from Li atoms, and two H2 molecules escape from the binding of the Li atoms at 250 K. At 300 K, nine H2 molecules can be stably absorbed on both sides of Li-PG, and the gravimetric hydrogen storage capacity can reach up to 9.25 wt.%, which is much higher than the the US Department of Energy target value of 5.5 wt.% for the year 2017. With the increase of temperature, more adsorbed H2 molecules are desorbed, seven H2 molecules can be desorbed at 400 K, and all H2 molecules are completely desorbed in a temperature range of 600–700 K.
-
Keywords:
- porous graphene /
- hydrogen storage /
- first-principles /
- molecular-dynamic
[1] Jiang L L, Fan Z J 2014 Nanoscale 6 1922
 Google Scholar
Google Scholar
[2] Liu W, Wang Z F, Shi Q W, Yang J L, Liu F 2009 Phys. Rev. B 80 233405
Google Scholar
[3] Bieri M, Treier M, Cai J M, Aït-Mansour K, Ruffieux P, Gröning O, Gröning P, Kastler M, Rieger R, Feng X L, Müllen K, Fasel R 2009 Chem. Commun. 45 6919
[4] Du A J, Zhu Z H, Smith S C 2010 J. Am. Chem. Soc. 132 2876
Google Scholar
[5] Li Y F, Zhou Z, Shen P W, Chen Z F 2010 Chem. Commun. 46 3672
Google Scholar
[6] Hatanaka M 2010 Chem. Phys. Lett. 488 187
Google Scholar
[7] Jiang D E, Cooper V R, Dai S 2009 Nano Lett. 9 4019
Google Scholar
[8] Merchant C A, Healy K, Wanunu M, Ray V, Petermn N, Battel J, Fischbein M D, Venta K, Luo Z T, Johnson C, Drndić M 2010 Nano Lett. 10 2915
Google Scholar
[9] Mukherjee R, Thomas A V, Datta D, Singh E, Li J W, Eksik O, Shenoy V B, Koratkar N 2014 Nature Commun. 5 3710
Google Scholar
[10] Bi H, Xie X, Yin K, Zhou Y, Wan S, He L, Xu F, Banhart F, Sun L, Ruoff R S 2012 Adv. Funct. Mater 22 4421
Google Scholar
[11] Reunchan P, Jhi S H 2011 Appl. Phys. Lett. 98 093103
Google Scholar
[12] Ao Z M, Dou S X, Xu Z M, Jiang Q G, Wang G X 2014 Int. J. Hydrogen energy 39 16244
Google Scholar
[13] Yuan L H, Chen Y H, Kang L, Zhang C R, Wang D B, Wang C N, Zhang M L, Wu X J 2017 Appl. Surf. Sci. 399 463
Google Scholar
[14] Yuan L H, Kang L, Chen Y H, Wang D B, Gong J J, Wang C N, Zhang M L, Wu X J 2018 Appl. Surf. Sci. 434 843
Google Scholar
[15] Yuan L H, Wang D B, Gong J J, Zhang C R, Zhang L P, Zhang M L, Wu X J, Kang L 2019 Chem. Phys. Lett. 726 57
Google Scholar
[16] Lu R F, Rao D W, Lu Z L, Qian J C, Li F, Wu H P, Wang Y Q, Xiao C Y, Deng K M, Kan E J, Deng W Q 2012 J. Phys. Chem. C 116 21291
Google Scholar
[17] Huang S H, Miao L 2012 J. Appl. Phys. 112 124312
Google Scholar
[18] Wang Y, Ji Y, Li M, Yuan P, Sun Q, Jia Y 2011 J. App. Phys. 110 094311
Google Scholar
[19] Chen Y D, Yu S, Zhao W H, Li S F, Duan X M 2018 Phys. Chem. Chem. Phys. 20 13473
Google Scholar
[20] Wang F D, Zhang T, Hou X Y, Zhang W Q, Tang S W, Sun H, Zhang J P 2017 Int. J. Hydrogen Energy 42 10099
Google Scholar
[21] Hashmi A, Farooq M U, Khan I, Son J, Hong J 2017 J. Mater. Chem. A 5 2821
Google Scholar
[22] Ning G Q, Xu C G, Mu L, Chen G J, Wang G, Gao J S, Fan Z J, Qian W Z, Wei F 2012 Chem. Commun. 48 6815
Google Scholar
[23] Xia K S, Tian X L, Fei S X, You K 2014 Int. J. Hydrogen Energy 39 11047
Google Scholar
[24] Elyassi M, Rashidi A, Hantehzadeh M R, Elahi S M 2017 Surf. Interface Anal 49 230
Google Scholar
[25] Vanderbilt D 1990 Phys. Rev. B 41 7892
Google Scholar
[26] Perdew J P, Burke K, Ernzerhof M 1996 Phys. Rev. Lett. 77 3865
Google Scholar
[27] Ao Z M, Peeters F M 2010 Phys. Rev. B 81 205406
Google Scholar
[28] Rao D W, Lu R F, Meng Z S, Wang Y, Lu Z L, Liu Y Z, Chen X, Kan E, Xiao C Y, Deng K M, Wu H P 2014 Int.J. Hydrogen Energy 39 18966
Google Scholar
[29] 基泰尔 C 著 (项金钟, 吴兴惠 译) 2009 固体物理导论 (北京: 化学工业出版社) 第38页
Kittel C (translated by Xiang J Z, Wu X H) 2009 Introduction to Solid State Physics (Beijing: Chemical Industry Press) p38 (in Chinese)
[30] 周晓锋, 方浩宇, 唐春梅 2019 物理学报 68 053601
Google Scholar
Zhou X F, Fang H Y, Tang C M 2019 Acta Phys. Sin. 68 053601
Google Scholar
[31] Seentithurai S, Kodi Pandya R, Vinodh Kumar S, Saranya C, mahendran M 2014 Int. J. Hydrogen Energy 39 11016
Google Scholar
[32] Sacchi M, Jenkins S J 2014 Phys. Chem. Chem. Phys. 16 6101
Google Scholar
-
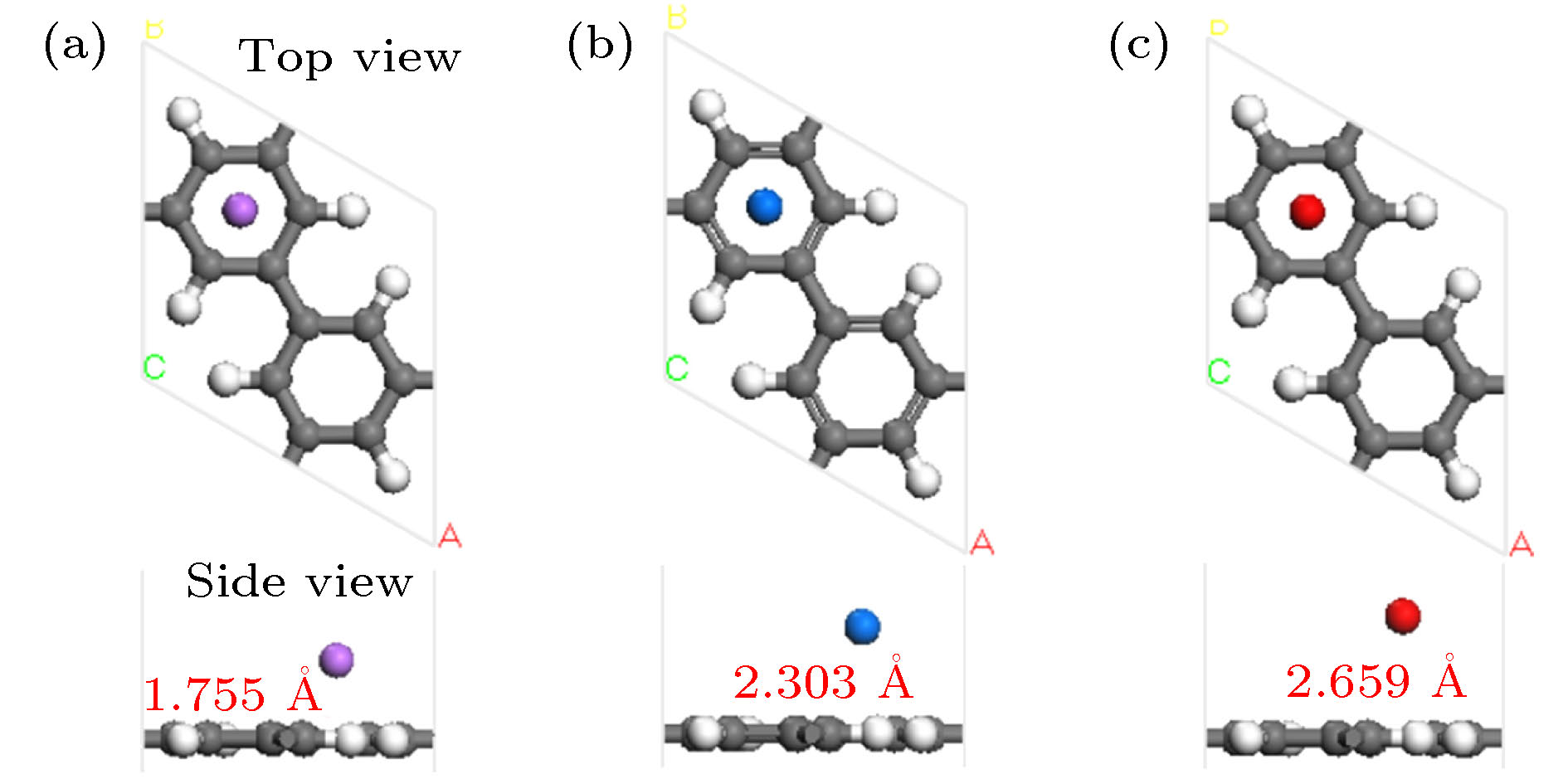
图 1 一个碱金属原子修饰PG优化后的几何结构 (a) Li-PG; (b) Na-PG; (c) K-PG
Fig. 1. Optimized geometry structure of a alkaline metal atom decorated PG: (a) Li-PG; (b) Na-PG; (c) K-PG.
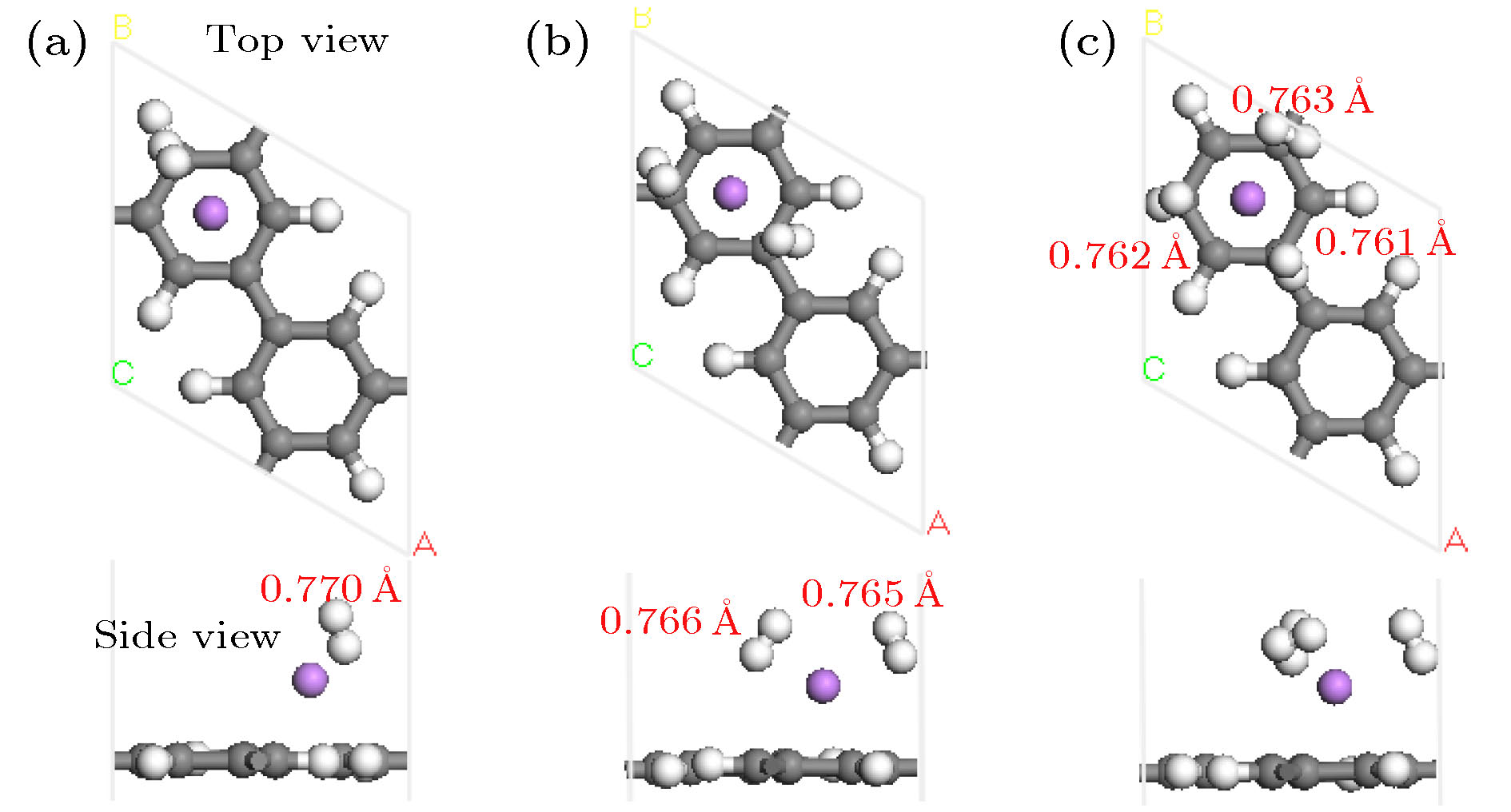
图 2 Li-PG吸附氢气分子弛豫后的几何结构(红色字体表示H—H键长)
Fig. 2. Optimized geometry structure of the Li-PG with H2 molecules adsorption. Red digits represent the corresponding bond length of H—H.
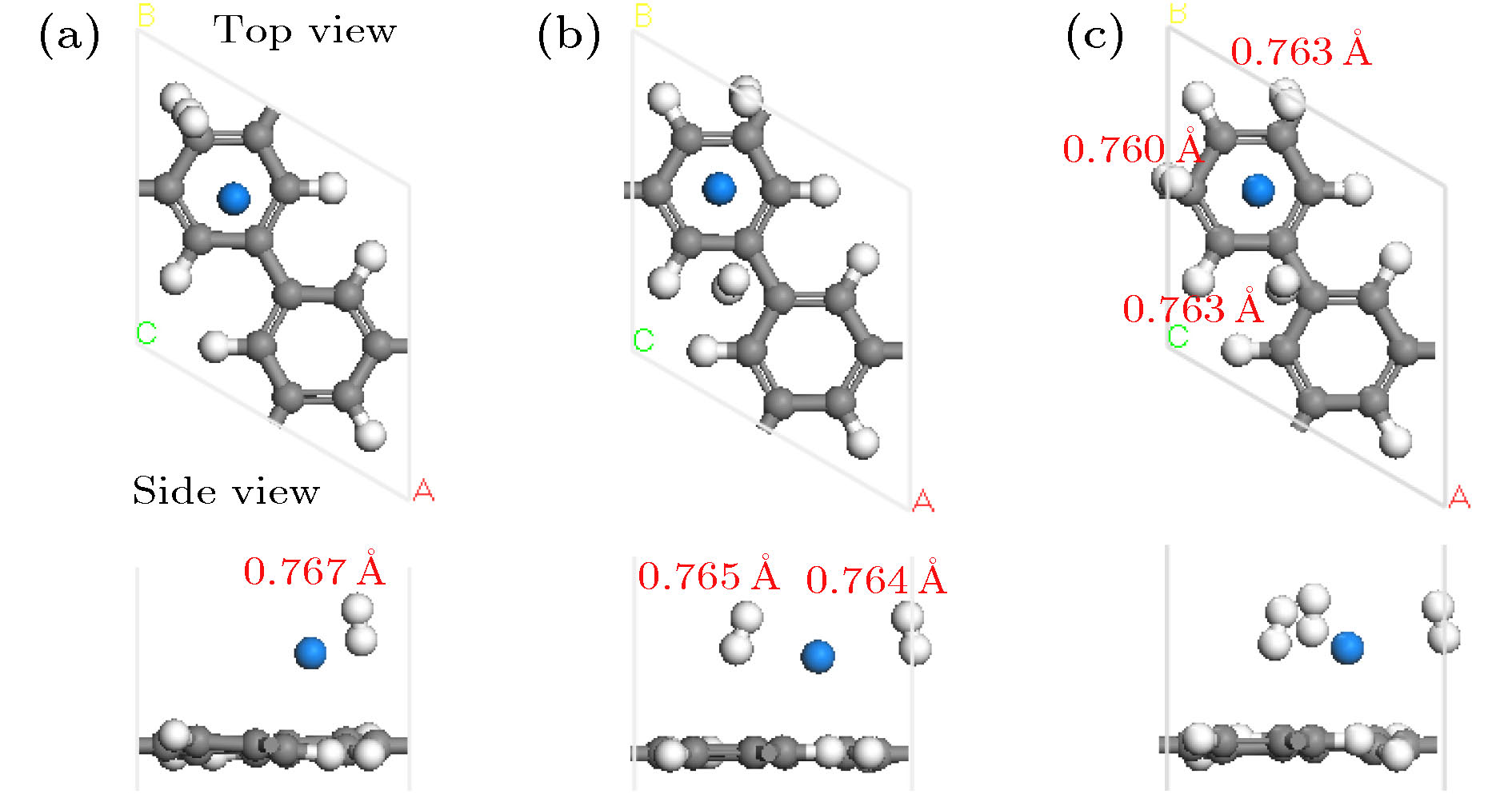
图 3 Na-PG吸附H2分子优化后的几何结构图(红色数字表示H—H键长)
Fig. 3. Optimized geometry structure of the Na-PG with H2 molecules adsorption. Red digits represent the corresponding bond length of H—H
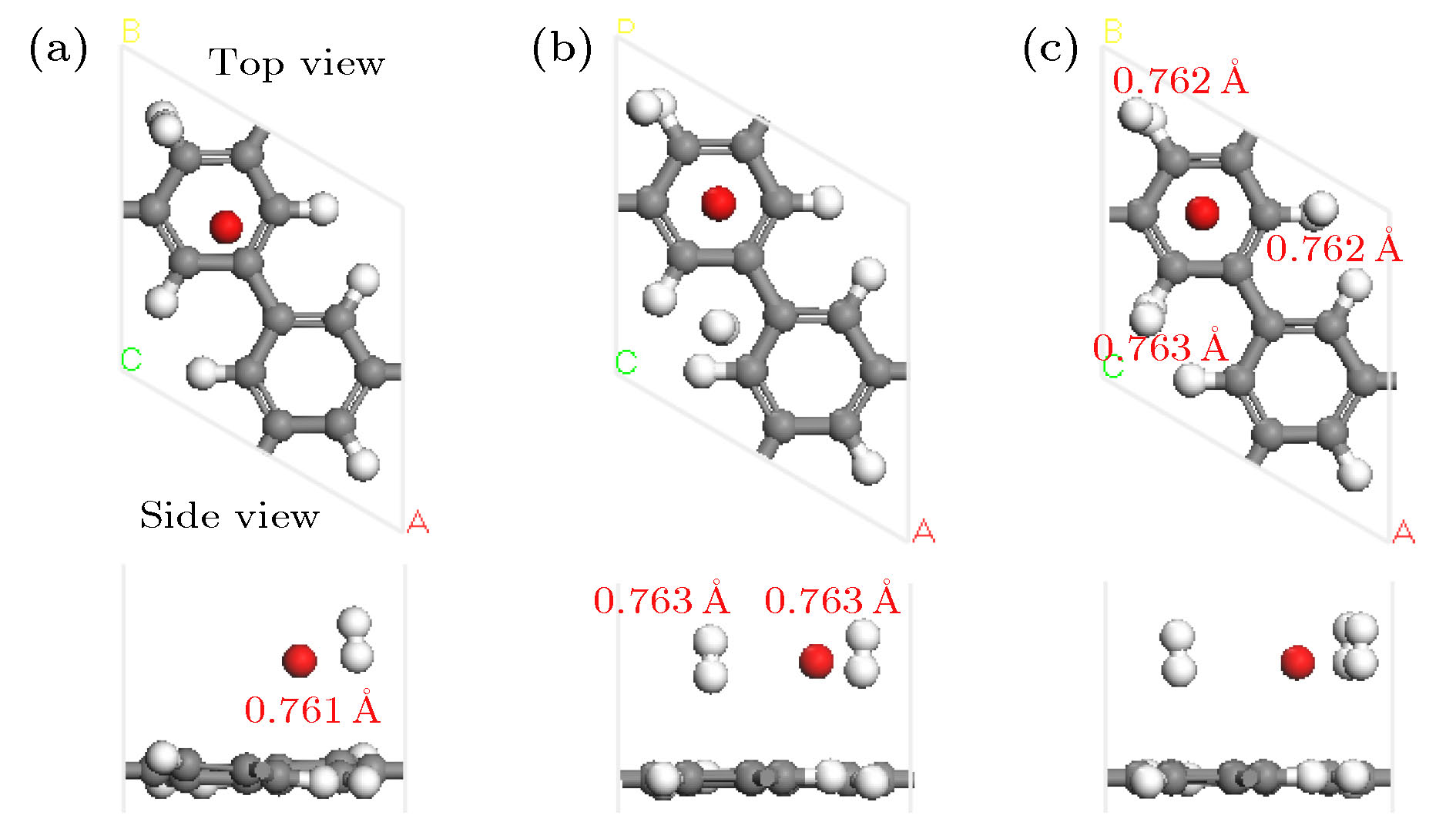
图 4 K-PG结构吸附H2分子优化后的几何结构图(红色数字表示H—H键长)
Fig. 4. Optimized geometry structure of the K-PG with H2 molecules. Red digits represent the corresponding bond length of H—H
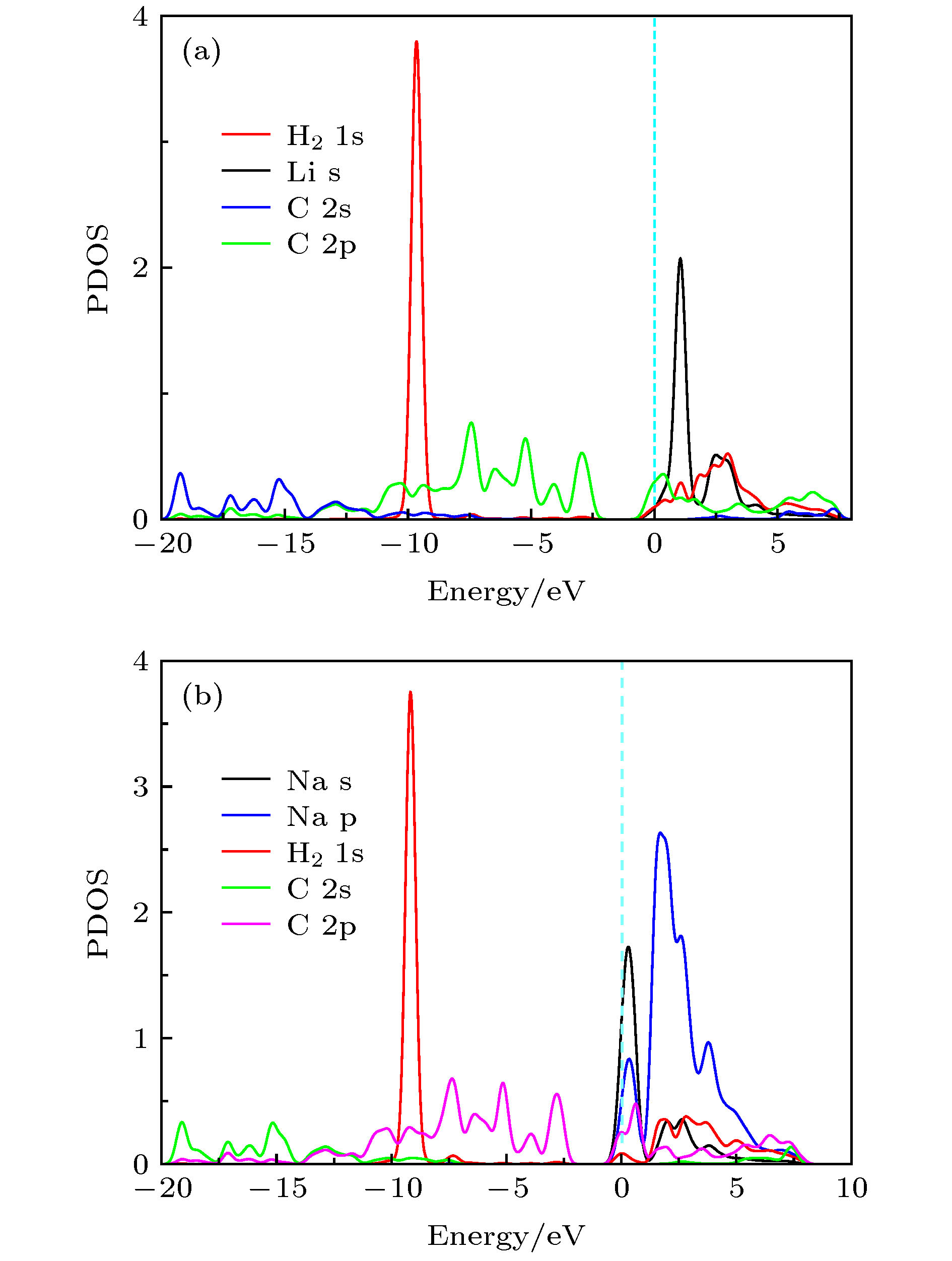
图 5 (a) Li-PG和(b) Na-PG结构吸附一个H2分子的分波态密度图
Fig. 5. Partial density of states (PDOS) of a H2 molecule on (a) Li-PG and (b) Na-PG.
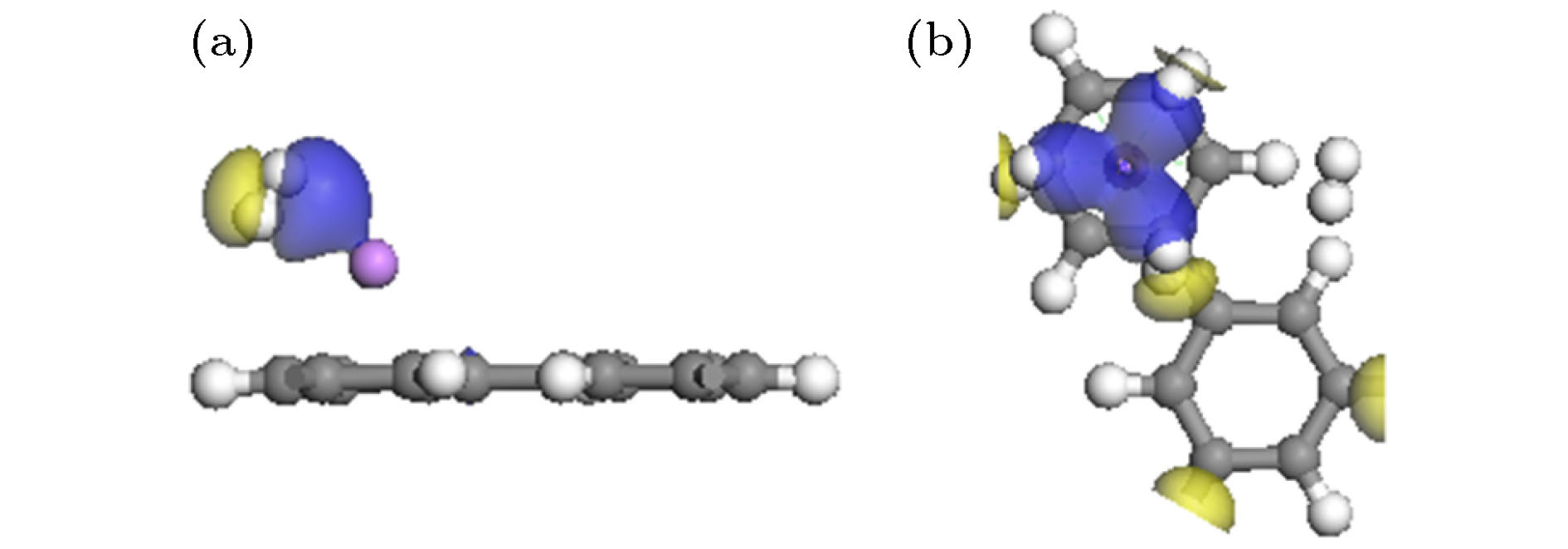
图 6 Li-PG吸附H2分子的差分电荷密度图(电荷密度等值面是0.01 e / Å3) (a) n = 1; (b) n = 4
Fig. 6. Charge density differences of n H2 adsorbed on Li-PG system for (a) n = 1 and (b) n = 4. The isovalue is taken to be 0.01 e/Å3
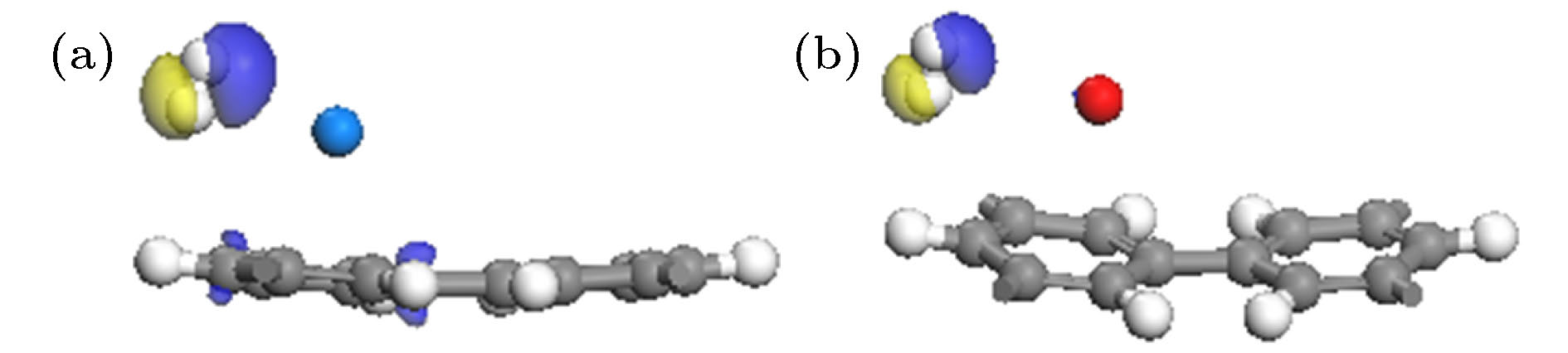
图 7 Na-PG (a)及K-PG (b)吸附1个H2的差分电荷密度图(电荷密度等值面是0.01 e/Å3)
Fig. 7. Charge density differences of a H2 adsorbed on Na-PG (a) and K-PG (b) system. The isovalue is taken to be 0.01 e/Å3.
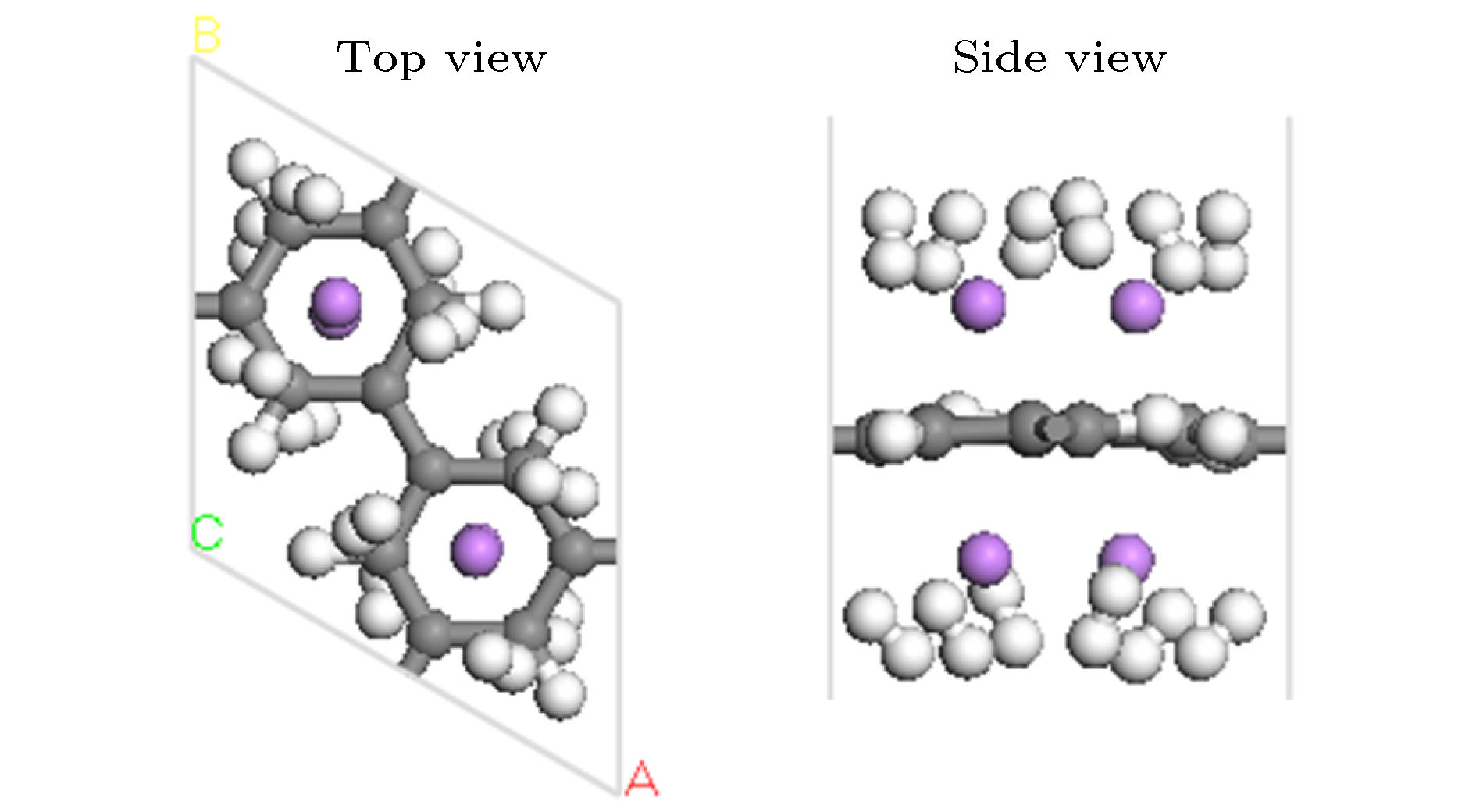
图 8 Li-PG结构吸附12个H2分子优化后的几何结构图
Fig. 8. Optimized geometry structure of the Li-decorated PG with 12 H2 adsorption.

图 9 经过AIMD模拟后12 H2-Li-PG平衡结构(a)及总能随模拟时间变化的函数关系(b)
Fig. 9. Equilibrated structure of the 12 H2-Li-PG (a) and fluctuations of total energy as a function of simulation time (b) in AIMD simulations at 300 K.
表 1 H2吸附能(ΔEad)、平均吸附能(
$\Delta {\bar E_{{\rm{ad}}}}$ )及Li所带的电荷(q)Table 1. Adsorption energies (ΔEad) and average adsorption energies (
$\Delta {\bar E_{{\rm{ad}}}}$ )of H2 molecules, and the charge of Li (q).n(H2) 1 2 3 4 ΔEad /eV –0.308 –0.239 –0.249 –0.044 $\Delta {\bar E_{{\rm{ad}}}}$/eV·H2–1 –0.308 –0.274 –0.266 –0.225 q /e 1.23 1.40 1.56 1.56  下载: 导出CSV
下载: 导出CSV
表 2 Na-PG吸附H2分子的吸附能
Table 2. Adsorption energies of H2 molecules on Na-PG.
n(H2) 1 2 3 ΔEh1/eV –0.202 –0.262 –0.215 ΔEh2/eV –0.234 –0.117 –0.166
下载: 导出CSV
-
[1] Jiang L L, Fan Z J 2014 Nanoscale 6 1922
Google Scholar
[2] Liu W, Wang Z F, Shi Q W, Yang J L, Liu F 2009 Phys. Rev. B 80 233405
Google Scholar
[3] Bieri M, Treier M, Cai J M, Aït-Mansour K, Ruffieux P, Gröning O, Gröning P, Kastler M, Rieger R, Feng X L, Müllen K, Fasel R 2009 Chem. Commun. 45 6919
[4] Du A J, Zhu Z H, Smith S C 2010 J. Am. Chem. Soc. 132 2876
Google Scholar
[5] Li Y F, Zhou Z, Shen P W, Chen Z F 2010 Chem. Commun. 46 3672
Google Scholar
[6] Hatanaka M 2010 Chem. Phys. Lett. 488 187
Google Scholar
[7] Jiang D E, Cooper V R, Dai S 2009 Nano Lett. 9 4019
Google Scholar
[8] Merchant C A, Healy K, Wanunu M, Ray V, Petermn N, Battel J, Fischbein M D, Venta K, Luo Z T, Johnson C, Drndić M 2010 Nano Lett. 10 2915
Google Scholar
[9] Mukherjee R, Thomas A V, Datta D, Singh E, Li J W, Eksik O, Shenoy V B, Koratkar N 2014 Nature Commun. 5 3710
Google Scholar
[10] Bi H, Xie X, Yin K, Zhou Y, Wan S, He L, Xu F, Banhart F, Sun L, Ruoff R S 2012 Adv. Funct. Mater 22 4421
Google Scholar
[11] Reunchan P, Jhi S H 2011 Appl. Phys. Lett. 98 093103
Google Scholar
[12] Ao Z M, Dou S X, Xu Z M, Jiang Q G, Wang G X 2014 Int. J. Hydrogen energy 39 16244
Google Scholar
[13] Yuan L H, Chen Y H, Kang L, Zhang C R, Wang D B, Wang C N, Zhang M L, Wu X J 2017 Appl. Surf. Sci. 399 463
Google Scholar
[14] Yuan L H, Kang L, Chen Y H, Wang D B, Gong J J, Wang C N, Zhang M L, Wu X J 2018 Appl. Surf. Sci. 434 843
Google Scholar
[15] Yuan L H, Wang D B, Gong J J, Zhang C R, Zhang L P, Zhang M L, Wu X J, Kang L 2019 Chem. Phys. Lett. 726 57
Google Scholar
[16] Lu R F, Rao D W, Lu Z L, Qian J C, Li F, Wu H P, Wang Y Q, Xiao C Y, Deng K M, Kan E J, Deng W Q 2012 J. Phys. Chem. C 116 21291
Google Scholar
[17] Huang S H, Miao L 2012 J. Appl. Phys. 112 124312
Google Scholar
[18] Wang Y, Ji Y, Li M, Yuan P, Sun Q, Jia Y 2011 J. App. Phys. 110 094311
Google Scholar
[19] Chen Y D, Yu S, Zhao W H, Li S F, Duan X M 2018 Phys. Chem. Chem. Phys. 20 13473
Google Scholar
[20] Wang F D, Zhang T, Hou X Y, Zhang W Q, Tang S W, Sun H, Zhang J P 2017 Int. J. Hydrogen Energy 42 10099
Google Scholar
[21] Hashmi A, Farooq M U, Khan I, Son J, Hong J 2017 J. Mater. Chem. A 5 2821
Google Scholar
[22] Ning G Q, Xu C G, Mu L, Chen G J, Wang G, Gao J S, Fan Z J, Qian W Z, Wei F 2012 Chem. Commun. 48 6815
Google Scholar
[23] Xia K S, Tian X L, Fei S X, You K 2014 Int. J. Hydrogen Energy 39 11047
Google Scholar
[24] Elyassi M, Rashidi A, Hantehzadeh M R, Elahi S M 2017 Surf. Interface Anal 49 230
Google Scholar
[25] Vanderbilt D 1990 Phys. Rev. B 41 7892
Google Scholar
[26] Perdew J P, Burke K, Ernzerhof M 1996 Phys. Rev. Lett. 77 3865
Google Scholar
[27] Ao Z M, Peeters F M 2010 Phys. Rev. B 81 205406
Google Scholar
[28] Rao D W, Lu R F, Meng Z S, Wang Y, Lu Z L, Liu Y Z, Chen X, Kan E, Xiao C Y, Deng K M, Wu H P 2014 Int.J. Hydrogen Energy 39 18966
Google Scholar
[29] 基泰尔 C 著 (项金钟, 吴兴惠 译) 2009 固体物理导论 (北京: 化学工业出版社) 第38页
Kittel C (translated by Xiang J Z, Wu X H) 2009 Introduction to Solid State Physics (Beijing: Chemical Industry Press) p38 (in Chinese)
[30] 周晓锋, 方浩宇, 唐春梅 2019 物理学报 68 053601
Google Scholar
Zhou X F, Fang H Y, Tang C M 2019 Acta Phys. Sin. 68 053601
Google Scholar
[31] Seentithurai S, Kodi Pandya R, Vinodh Kumar S, Saranya C, mahendran M 2014 Int. J. Hydrogen Energy 39 11016
Google Scholar
[32] Sacchi M, Jenkins S J 2014 Phys. Chem. Chem. Phys. 16 6101
Google Scholar



 下载:
下载:


计量
- 文章访问数: 14224
- PDF下载量: 170
- 被引次数: 0
