










-
原子表征与操控是实现原子制造必须突破的物理瓶颈之一. 像差校正电子显微学方法因其优异的空间分辨率, 为实现原子精细制造提供了有力的表征手段. 因此, 利用电子显微学手段, 在原子尺度对原子制造的材料及器件进行三维结构和性能的协同表征, 对于深入理解原子水平材料操控的物理机理具有非常重要的意义. 纳米团簇及纳米颗粒是原子制造材料与器件研究的主要对象之一, 具有丰富的物理化学性质和较高的可操纵性. 本文探讨纳米团簇/颗粒结构三维定量表征、使役条件下纳米团簇/颗粒结构演变定量表征、纳米颗粒/晶粒结构-成分-磁性协同定量表征等诸多方法与实例, 阐明了电子显微学表征手段的突破和发展为实现精细控制的原子制造材料提供了坚实基础.
-
关键词:
- 原子制造 /
- 透射电子显微学与谱学 /
- 三维结构定量表征 /
- 纳米团簇
Atomic scale characterization and manipulation is one of the physical bottlenecks, which needs to be broken when realizing atom manufacturing. The aberration-corrected transmission electron microscopy (TEM) is a powerful tool for structural characterization due to its exceptional spatial resolution. Therefore, it is very crucial to co-characterize atomic-scale three-dimensional structure and properties of atomic manufacturing materials by using TEM, which allows us to further understand the physics mechanism of atomic manipulation of materials. Nano-clusters and nanoparticles are two of the main objects in the studies of atomic manufacturing materials and devices, and possess rich physical and chemical properties and high manoeuverability. In this paper, we summarize the recent progress of quantitatively determining three-dimensional structures and magnetic properties of nanocluster, nanoparticles and nanograins, as well as their dynamic evolutions under the working conditions. The methodological breakthrough and development of electron microscopy techniques provide a solid foundation for precisely controlling atomic manufacturing materials.-
Keywords:
- atom manufacturing /
- transmission electron microscopy and spectroscopy /
- three-dimensional quantitative structure characterization /
- nanoclusters
[1] Cai R, Martelli F, Vernieres J, Albonetti S, Dimitratos N, Tizaoui C, Palmer R E 2020 ACS Appl. Mater. Interfaces 12 24877
 Google Scholar
Google Scholar
[2] Curl R F, Smalley R E 1991 Sci. Am. 265 54
[3] Kroto H W, Heath J R, O’Brien S C, Curl R F, Smalley R E 1985 Nature 318 162
Google Scholar
[4] Becker E W, Bier K, Henkes W 1956 Z. Physik 146 333
Google Scholar
[5] Purusottam J, A Welford C Jr 2010 Nanoclusters: a Bridge Across Disciplines (Oxford: Elsevier) pp3−36
[6] Khanna S N, Jena P 1992 Phys. Rev. Lett. 69 1664
Google Scholar
[7] Zhou J, Yang Y, Ercius P, Miao J 2020 MRS Bull. 45 290
Google Scholar
[8] Miller M K 2000 Atom Probe Tomography: Analysis at the Atomic Level (New York: Springer) pp1−23
[9] Midgley P A, Weyland M 2003 Ultramicroscopy 96 413
Google Scholar
[10] Haider M, Uhlemann S, Schwan E, Rose H, Kabius B, Urban K 1998 Nature 392 768
Google Scholar
[11] Midgley P A, Dunin Borkowski R E 2009 Nat. Mater. 8 271
Google Scholar
[12] Urban K W 2009 Nat. Mater. 8 260
Google Scholar
[13] Muller D A 2009 Nat. Mater. 8 263
Google Scholar
[14] Chen F R, Van Dyck D, Kisielowski C 2016 Nat. Commun. 7 10603
Google Scholar
[15] Wiesendanger R, Shvets I V, Bürgler D, Tarrach G, Güntherodt H J, Coey J M, Gräser S 1992 Science 255 583
Google Scholar
[16] Heinze S, Bode M, Kubetzka A, Pietzsch O, Nie X, Blügel S, Wiesendanger R 2000 Science 288 1805
Google Scholar
[17] Kaiser U, Schwarz A, Wiesendanger R 2007 Nature 446 522
Google Scholar
[18] Beaurepaire E, Kappler J P, Krill G, Scheurer F 2010 Magnetism and Synchrotron Radiation (Berlin: Springer) pp145−364
[19] Chao W, Harteneck B, Liddle J, Anderson E, Attwood D 2005 Nature 435 1210
Google Scholar
[20] Kim D H, Fischer P, Chao W, Anderson E, Im M Y, Shin S, Choe S B 2006 J. Appl. Phys. 99 08H303
Google Scholar
[21] Zhu X, Hitchcock A P, Bazylinski D A, Denes P, Joseph J, Lins U, Marchesini S, Shiu H W, Tyliszczak T, Shapiro D A 2016 Proc. Natl. Acad. Sci. U. S. A. 113 E8219
Google Scholar
[22] Pennycook S J 2011 Scanning Transmission Electron Microscopy: Imaging and Analysis (New York: Springer) pp1−90
[23] Jia C, Lentzen M, Urban K 2003 Science 299 870
Google Scholar
[24] Li Z Y, Young N P, Di Vece M, Palomba S, Palmer R E, Bleloch A L, Curley B C, Johnston R L, Jiang J, Yuan J 2008 Nature 451 46
Google Scholar
[25] Bals S, Van Aert S, Romero C P, Lauwaet K, Van Bael M J, Schoeters B, Partoens B, Yücelen E, Lievens P, Van Tendeloo G 2012 Nat. Commun. 3 897
Google Scholar
[26] Lentzen M, Jahnen B, Jia C L, Thust A, Tillmann K, Urban K 2002 Ultramicroscopy 92 233
Google Scholar
[27] Jia C L, Mi S B, Barthel J, Wang D W, Dunin Borkowski R E, Urban K W, Thust A 2014 Nat. Mater. 13 1044
Google Scholar
[28] Van Dyck D, Jinschek J R, Chen F R 2012 Nature 486 243
Google Scholar
[29] Hawkes P W 1980 Computer Processing of Electron Microscope Images (Berlin: Springer) pp1−33
[30] Kirkland E J 1984 Ultramicroscopy 15 151
Google Scholar
[31] Op de Beeck M, Van Dyck D, Coene W 1996 Ultramicroscopy 64 167
Google Scholar
[32] Meyer R R, Kirkland A I, Saxton W O 2002 Ultramicroscopy 92 89
Google Scholar
[33] Allen L J, McBride W, O'Leary N L, Oxley M P 2004 Ultramicroscopy 100 91
Google Scholar
[34] Reich S, Maultzsch J, Thomsen C, Ordejón P 2002 Phys. Rev. B 66 035412
Google Scholar
[35] Miedema M A O, van den Bos A, Buist A H 1994 IEEE Instrum. Meas. Technol. Conf. 43 181
Google Scholar
[36] Buist A H, van den Bos A, Miedema M A O 1996 Ultramicroscopy 64 137
Google Scholar
[37] Iijima S, Ichihashi T 1986 Phys. Rev. Lett. 56 616
Google Scholar
[38] Ajayan P, Marks L 1989 Phys. Rev. Lett. 63 279
Google Scholar
[39] Smith D J, Petford Long A K, Wallenberg L R, Bovin J O 1986 Science 233 872
Google Scholar
[40] Van Dyck D, Lobato I, Chen F R, Kisielowski C 2015 Micron 68 158
Google Scholar
[41] De Rosier D J, Klug A 1968 Nature 217 130
Google Scholar
[42] Hoppe W, Langer R, Knesch G, Poppe C 1968 Naturwissenschaften 55 333
Google Scholar
[43] Hart R G 1968 Science 159 1464
Google Scholar
[44] Arslan I, Yates T J V, Browning N D, Midgley P A 2005 Science 309 2195
Google Scholar
[45] Xia Y, Zhong X Y, Ke X, Zhang G R, Cheng Z, Xu B Q 2016 Small 12 6332
Google Scholar
[46] Zhong X Y, Kabius B, Schreiber D, Eastman J, Fong D, Petford Long A 2012 Appl. Phys. Lett. 100 101604
Google Scholar
[47] Zhong X Y, Kabius B, Schreiber D K, Eastman J A, Fong D D, Petford Long A K 2009 Microsc. microanal. 15 600
Google Scholar
[48] Van Aert S, Batenburg K J, Rossell M D, Erni R, Van Tendeloo G 2011 Nature 470 374
Google Scholar
[49] Miao J W, Ercius P, Billinge S J L 2016 Science 353 aaf2157
Google Scholar
[50] Scott M C, Chen C C, Mecklenburg M, Zhu C, Xu R, Ercius P, Dahmen U, Regan B C, Miao J W 2012 Nature 483 444
Google Scholar
[51] Chen C C, Zhu C, White E R, Chiu C Y, Scott M C, Regan B C, Marks L D, Huang Y, Miao J W 2013 Nature 496 74
Google Scholar
[52] Xu R, Chen C C, Wu L, Scott M C, Theis W, Ophus C, Bartels M, Yang Y, Ramezani Dakhel H, Sawaya M R, Heinz H, Marks L D, Ercius P, Miao J W 2015 Nat. Mater. 14 1099
Google Scholar
[53] Yang Y, Chen C C, Scott M C, Ophus C, Xu R, Pryor A, Wu L, Sun F, Theis W, Zhou J, Eisenbach M, Kent P R C, Sabirianov R F, Zeng H, Ercius P, Miao J 2017 Nature 542 75
Google Scholar
[54] Haruta M, Kobayashi T, Sano H, Yamada N 1987 Chem. Lett. 16 405
Google Scholar
[55] Foster D M, Ferrando R, Palmer R E 2018 Nat. Commun. 9 1323
Google Scholar
[56] Foster D M, Pavloudis T, Kioseoglou J, Palmer R E 2019 Nat. Commun. 10 2583
Google Scholar
[57] Zhou J, Yang Y, Yang Y, Kim D S, Yuan A, Tian X, Ophus C, Sun F, Schmid A K, Nathanson M, Heinz H, An Q, Zeng H, Ercius P, Miao J 2019 Nature 570 500
Google Scholar
[58] Zhu B, Meng J, Yuan W, Zhang X, Yang H, Wang Y, Gao Y 2020 Angew. Chem. Int. Edit. 59 2171
Google Scholar
[59] Altantzis T, Lobato I, De Backer A, Béché A, Zhang Y, Basak S, Porcu M, Xu Q, Sánchez Iglesias A, Liz Marzán L M, Van Tendeloo G, Van Aert S, Bals S 2019 Nano Lett. 19 477
Google Scholar
[60] Abrams I M, McBain J W 1944 J. Appl. Phys. 15 607
Google Scholar
[61] Swift J A, Brown A C 1970 J. Phys. E 3 924
Google Scholar
[62] Zheng H, Smith R, Jun Y W, Kisielowski C, Dahmen U, Alivisatos A 2009 Science 324 1309
Google Scholar
[63] Park J, Zheng H, Lee W C, Geissler P L, Rabani E, Alivisatos A P 2012 ACS Nano 6 2078
Google Scholar
[64] Yuk J M, Park J, Ercius P, Kim K, Hellebusch D, Crommie M, Lee J, Zettl A, Alivisatos A 2012 Science 336 61
Google Scholar
[65] Liao H G, Zherebetskyy D, Xin H, Czarnik C, Ercius P, Elmlund H, Pan M, Wang L W, Zheng H 2014 Science 345 916
Google Scholar
[66] Park J, Elmlund H, Ercius P, Yuk J M, Limmer D T, Chen Q, Kim K, Han S H, Weitz D A, Zettl A, Alivisatos A P 2015 Science 368 290
Google Scholar
[67] Kim B H, Heo J, Kim S, Reboul C F, Chun H, Kang D, Bae H, Hyun H, Lim J, Lee H, Han B, Hyeon T, Alivisatos A P, Ercius P, Elmlund H, Park J 2020 Science 368 60
Google Scholar
[68] Sutter E, Jungjohann K, Bliznakov S, Courty A, Maisonhaute E, Tenney S, Sutter P 2014 Nat. Commun. 5 4946
Google Scholar
[69] Liu S Y, Kundu P, Huang T W, Chuang Y J, Tseng F G, Lu Y, Sui M L, Chen F R 2017 Nano Energy 31 218
Google Scholar
[70] Lu Y, Geng J, Wang K, Zhang W, Ding W, Zhang Z, Xie S, Dai H, Chen F R, Sui M 2017 ACS Nano 11 8018
Google Scholar
[71] Lu Y, Yin W J, Peng K L, Wang K, Hu Q, Selloni A, Chen F R, Liu L M, Sui M L 2018 Nat. Commun. 9 2752
Google Scholar
[72] Schattschneider P, Rubino S, Hébert C, Rusz J, Kuneš J, Novák P, Carlino E, Fabrizioli M, Panaccione G, Rossi G 2006 Nature 441 486
Google Scholar
[73] Wang Z Q, Zhong X Y, Yu R, Cheng Z Y, Zhu J 2013 Nat. Commun. 4 1395
Google Scholar
[74] Rusz J, Eriksson O, Novak P, Oppeneer P 2007 Phys. Rev. B 76 060408(R
Google Scholar
[75] Calmels L, Houdellier F, Warot Fonrose B, Gatel C, Hÿtch M, Serin V, Snoeck E, Schattschneider P 2007 Phys. Rev. B 76 060409(R
Google Scholar
[76] Lin J, Zhong X Y, Rusz J, Kocevski V, Xin H, Cui B, Han L, Lin R, Chen X, Zhu J 2017 Phys. Rev. Mater. 1 071404(R
Google Scholar
[77] Ho P L, Yu C P, Zhang Q, Song K, Buban J P, Choi S Y, Dunin Borkowski R E, Mayer J, Tai N H, Zhu J, Jin L, Zhong X Y 2018 Ultramicroscopy 193 137
Google Scholar
[78] Chen X, Higashikozono S, Ito K, Jin L, Ho P L, Yu C P, Tai N H, Mayer J, Dunin Borkowski R E, Suemasu T, Zhong X Y 2019 Ultramicroscopy 203 37
Google Scholar
[79] Schattschneider P, Stöger Pollach M, Rubino S, Sperl M, Hurm C, Zweck J, Rusz J 2008 Phys. Rev. B 78 104413
Google Scholar
[80] Thersleff T, Rusz J, Hjörvarsson B, Leifer K 2016 Phys. Rev. B 94 134430
Google Scholar
[81] Rusz J, Muto S, Spiegelberg J, Adam R, Tatsumi K, Bürgler D E, Oppeneer P M, Schneider C M 2016 Nat. Commun. 7 12672
Google Scholar
[82] Wang Z, Tavabi A H, Jin L, Rusz J, Tyutyunnikov D, Jiang H, Moritomo Y, Mayer J, Dunin Borkowski R E, Yu R, Zhu J, Zhong X Y 2018 Nat. Mater. 17 221
Google Scholar
[83] 陈鑫峰, 王泽朝, 钟虓䶮 2018 电子显微学报 37 540
Google Scholar
Chen X F, Wang Z C, Zhong X Y 2018 J. Chin. Electrn Microsc. Soc. 37 540
Google Scholar
[84] Almeida T P, Muxworthy A R, Kovács A, Williams W, Brown P D, Dunin Borkowski R E 2016 Sci. Adv. 2 e1501801
Google Scholar
[85] Almeida T P, Kasama T, Muxworthy A R, Williams W, Nagy L, Hansen T W, Brown P D, Dunin Borkowski R E 2014 Nat. Commun. 5 5154
Google Scholar
-
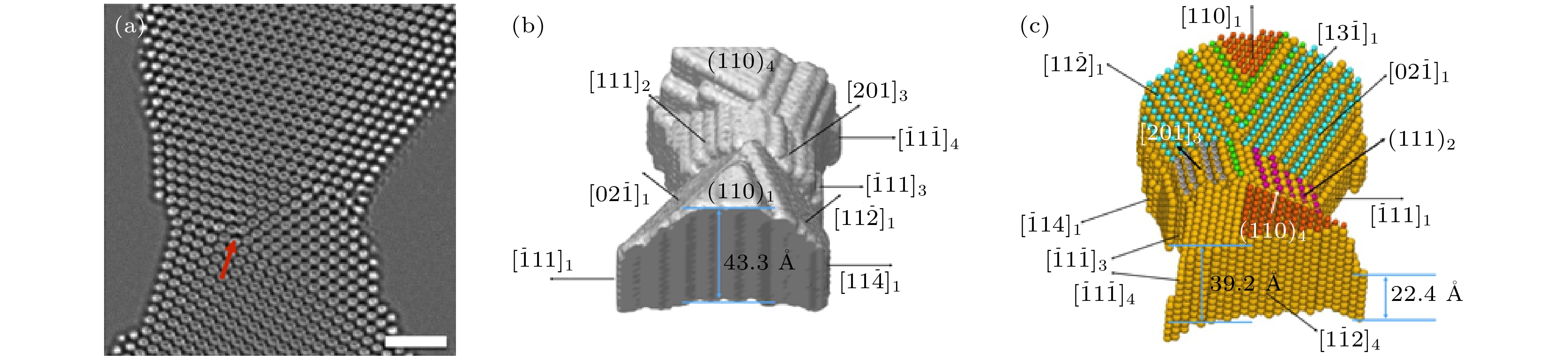
图 1 (a)加速电压为300 kV下的纳米金样品同轴全息图, 标尺为1 nm, 图中在3个孪晶的交接处用红色箭头标出了位错; 纳米金样品的三维(b)表面形貌和(c)原子构型, 并用不同的颜色标出不同晶体学取向的表面[14]
Fig. 1. (a) In-line holograms of a nanosized Au sample at the acceleration voltage of 300 kV. Scale bar is 1 nm. It is noteworthy that there are edge-on twin boundaries present in the imaged gold sample. At the intersection of three twin boundaries, a present end-on dislocation marked by the red arrow. (b) Surface morphology and (c) atomic structure views of the nanosized Au sample in three dimensions. The facets with different crystallographic orientations are highlighted with different colors[14].

图 2 原子级电子断层成像术示意图 (a)电子束聚焦在一个小点上并在样品上扫描以形成二维图像. 每个扫描位置的积分信号由环形暗场(ADF)探测器记录. (b)通过绕倾转轴旋转样品, 并在不同的倾转角度下测量一系列二维图像. (c)经过预处理和对中后, 通过分数傅里叶变换(FrFT)将系列倾转图像转换为傅里叶片层. 三维重构是通过傅里叶迭代算法来计算的. 通过三维重构, 可以跟踪和细化单个原子的坐标, 从而生成样品的三维原子模型[49]
Fig. 2. Schematic layout of atomic electron tomography (AET): (a) An electron beam is focused on a small spot and scanned over a sample to form a 2D image.The integrated signal at each scanning position is recorded by an annular dark-field (ADF) detector. (b) By rotating the sample around a tilt axis, a series of 2D images is measured at different tilt angles. (c) After preprocessing and alignment, the tilt series is inverted to Fourier slices by the fractional Fourier transform (FrFT). A 3D reconstruction is computed by using a Fourier-based iterative algorithm. From the 3D reconstruction, the coordinates of individual atoms are traced and refined to produce the 3D atomic model of the sample[49].

图 3 20 ℃和500 ℃下的Au561团簇的高角环形暗场扫描透射显微(HAADF STEM)图像 (a)—(d) Au561团簇HAADF STEM图像和(e)—(h)对应的不同带轴下立方八面体和Ino十面体的多片层法模拟像; (a), (b)在20 ℃下记录的实验像; (c), (d)在500 ℃下记录的实验像; (i)立方八面体和Ino十面体的几何构型及其旋转角度示意[55]
Fig. 3. High-angle annular dark-field scanning transmission electron microscopy (HAADF STEM) images of Au561 clusters at 20 ℃ and 500 ℃: (a)–(d) HAADF STEM images of Au561 clusters and (e)–(h) matching multi-slice electron scattering simulations of the cuboctahedron and Ino-decahedron at different orientations; (a), (b) experimental images recorded at 20 ℃; (c), (d) images recorded at 500 ℃; (i) rotation angle of the cuboctahedron and Ino-decahedron geometries[55].
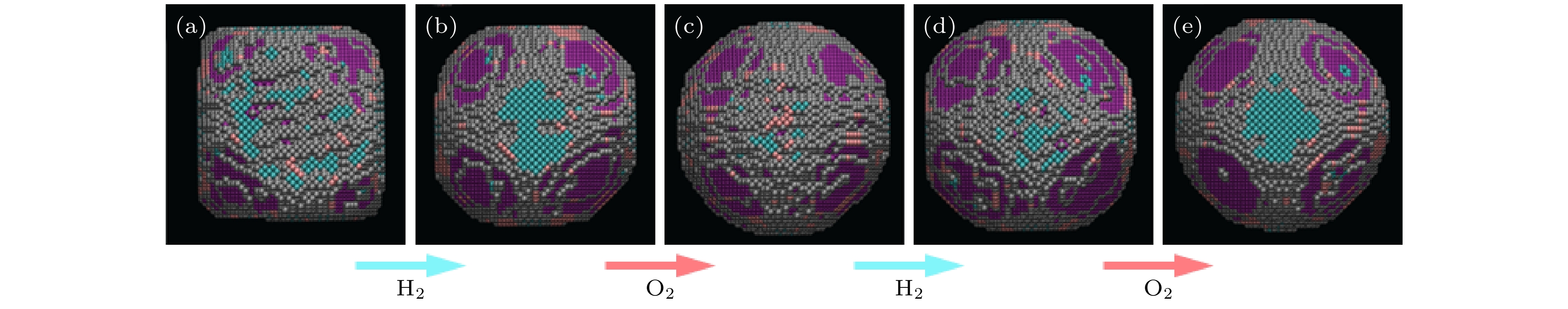
图 4 Pt纳米颗粒在不同条件下的三维结构演变形貌 (a) 在真空中; (b), (d) 300 ℃下通入含5% H2的Ar气; (c), (e) 300 ℃下通入O2; 颗粒表面上不同晶面族的原子用不同颜色表示, 蓝色对应{100}, 粉色对应{110}, 紫色对应{111}, 灰色对应高指数面[59]
Fig. 4. Three dimensional morphology of structural evolution of Pt nanoparticles under different environmental conditions: (a) In vacuum; (b), (d) in 5% H2 in Ar at 300 °C; (c), (e) in O2 at 300 °C. The atoms are presented in different colors, according to the type of surface facet: blue = {100}, pink = {110}, purple = {111}, gray = higher index[59].

图 5 使用原子级电子断层成像术表征FePt颗粒中四维原子运动 (a)—(c) FePt纳米颗粒在累计退火时间为9、6和26分钟时的三维原子模型(Fe是红色、Pt是蓝色); (d)—(f) 在三种不同退火时间下富Pt的核心保持不变, 模型下方的浅灰色和深灰色投影分别显示了整个纳米颗粒及其核心; (g)—(i) 在3个退火时间沿 [010] 方向相同的纳米颗粒内部原子构型(Fe原子为红色, Pt原子为蓝色), 其中表面原子和亚表面的一小部分原子重新排列形成L10相(由椭圆形标出), 标尺为1 nm; (j)—(l) 颗粒形核点在退火时间为9、16和26分钟的分布, 其中点的颜色越淡代表其离表面越近, 颜色越暗表明其越靠近内部,
$ \left\langle {100} \right\rangle $ 和$ \left\langle {111} \right\rangle $ 取向的刻面分别用绿色和品红色表示[67]Fig. 5. Capturing four-dimensional atomic motion with atomic electron tomography. (a)–(c) Three-dimensional atomic models (Fe in red and Pt in blue) of an FePt nanoparticle with an accumulated annealing time of 9, 16 and 26 min, respectively. (d)–(f) The Pt-rich core of the nanoparticle (shown here) remained the same for the three annealing times. The light and dark grey projections below the models show the whole nanoparticle and the core, respectively. (g)–(i) The same internal atomic layer of the nanoparticle along the [010] direction at the three annealing times (Fe in red and Pt in blue), where a fraction of the surface and subsurface atoms had rearranged to form L10 phase (ellipses). Scale bar is 1 nm. (j)–(l) Distribution of the nucleation sites (dots) in particle with an accumulated annealing time of 9, 16 and 26 min, respectively, where the lighter colored dots are closer to the front side and the darker dots are closer to the back side of the nanoparticle. The
$ \left\langle {100} \right\rangle $ and$ \left\langle {111} \right\rangle $ facets are in green and magenta, respectively[67].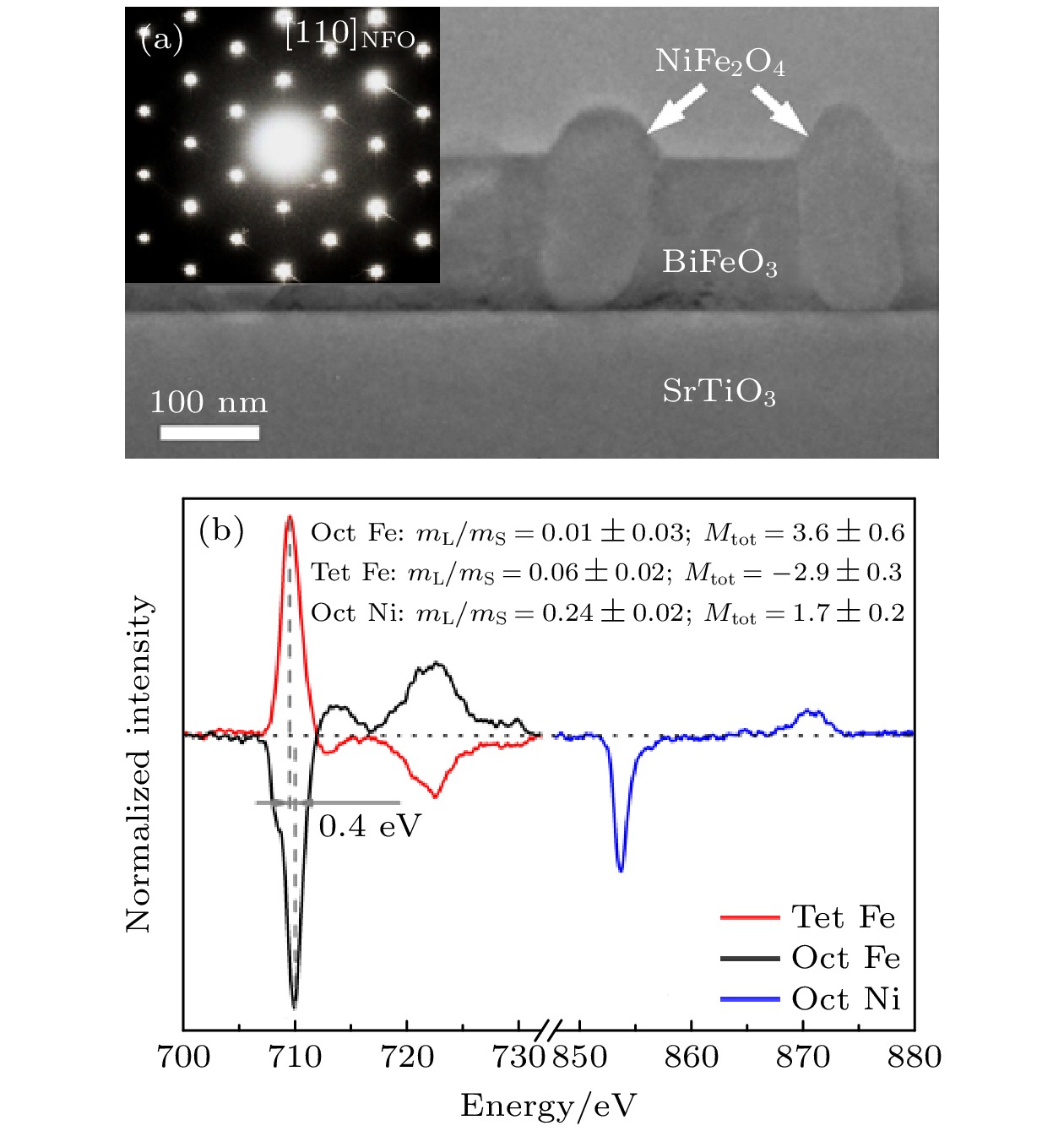
图 6 纳米尺度协同测量单一NiFe2O4纳米晶粒的晶体结构、元素成分与定量磁矩 (a) 复合薄膜截面样品的透射电子显微(TEM)图像和选区衍射 (SAED) 花样, 在SrTiO3基底上异质外延生长的钙钛矿BiFeO3纳米晶粒和尖晶石NiFe2O4纳米晶粒的截面样品的低倍TEM图像, NiFe2O4纳米晶粒柱在[110]带轴下的单晶SAED花样; (b) 八面体Fe (Oct Fe)、八面体Ni (Oct Ni)、四面体Fe (Tet Fe)的占位分辨的磁圆二色谱; Oct Fe和Tet Fe之间的0.4 eV的化学位移差别反映了不同占位对称性上的差异, mL/mS为轨道自旋磁矩比, MOct, Fe, MTet, Fe和MOct, Ni 是Oct Fe, Tet Fe和Oct Ni的总磁矩, 单位为μB/atom[73]
Fig. 6. Nanoscale co-located measurement of crystallographic structure, elemental composition and quantitative magnetic moments of single NiFe2O4 nanograin: (a) Transmission electron microscopy (TEM) image and selected area electron diffraction (SAED) patterns of cross-sectional composite films. Low-magnification TEM image for cross-section sample of hetero-epitaxial composite thin films formed by perovskite BiFeO3 and spinel NiFe2O4 nanograins on SrTiO3 substrates. The single-crystalline SAED patterns are acquired from one NiFe2O4 nanograin along the [110] zone axis. (b) Site-specific magnetic circular dichroism spectra for Fe atoms at octahedral sites (Oct Fe), Ni atoms at octahedral sites (Oct Ni) and Fe atoms at tetrahedral sites (Tet Fe). A chemical shift of 0.4 eV between oct Fe peak and Tet Fe peak reveals the chemical shift for different site symmetries. mL/mS refers to the orbital to spin magnetic moment ratio. MOct, Fe, MTet, Fe and MOct, Ni are the total magnetic moments (the sum of spin and orbital magnetic moments) of Oct Fe, Tet Fe and Oct Ni in the units of μB/atom[73].
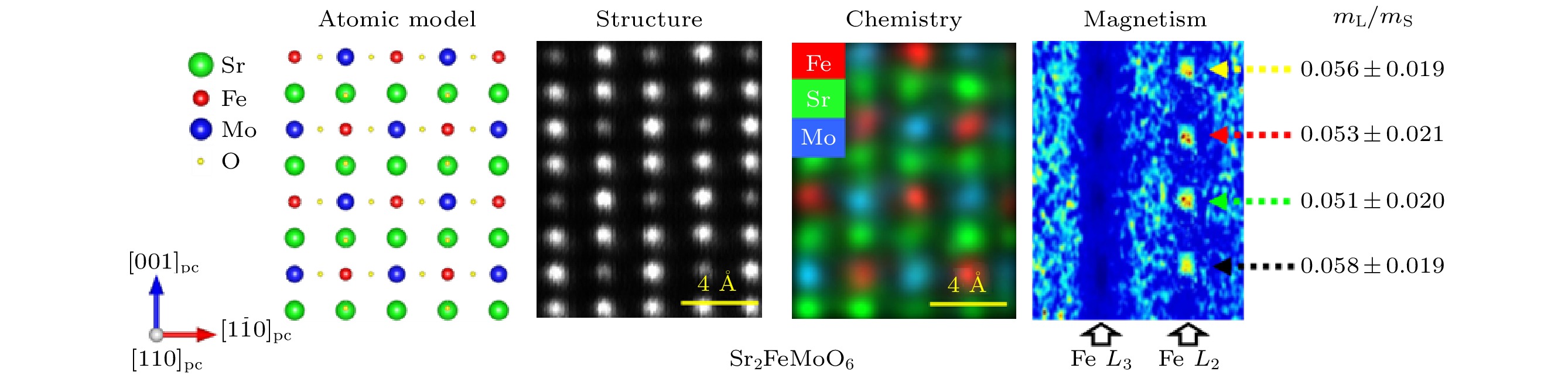

图 8 原位观测伪单畴的Fe3O4纳米颗粒的热磁行为 (a) 长对角轴的长度约为 150 nm的Fe3O4纳米颗粒的明场像以及对应的电子衍射花样; (b)—(h)依次在(b) 20 °C以及原位反应过程中加热到(c) 400 °C, (d) 500 °C和(e) 550 °C, 以及随后降温到 (f) 500 °C, (g) 400 °C和(h) 20 °C时记录的全息图重构出的磁感应强度分布图(标有FD字样的红色箭头代表诱导产生剩磁的外加饱和磁场的面内分量方向, 蓝色箭头表示为计算平均内势能而施加的额外的外磁场的方向); 所有磁感应强度分布图的等高磁轮廓线间距为 0.098 rad, 磁化方向使用箭头显示, 如色轮中所示; v代表漩涡的中心[84]
Fig. 8. Visualization of the thermomagnetic behavior of a small pseudo-single-domain Fe3O4 grain. (a) Bright-field transmission electron microscopy image of the individual Fe3O4 grain (about 150 nm in length across its long-diagonal axis), with associated electron diffraction pattern inset. (b)–(h) Magnetic induction maps reconstructed from holograms taken at (b) 20 °C (with the red arrow, labeled FD, showing the direction of the in-plane component of the applied saturating field to induce magnetic remanence, whereas the blue arrow shows the additional direction the field was applied for the calculation of the mean inner potential); during in situ heating to (c) 400 °C, (d) 500 °C, and (e) 550 °C; upon subsequent cooling to (f) 500 °C, (g) 400 °C, and (h) 20 °C. The contour spacing is 0.098 rad for all the magnetic induction maps, and the magnetization direction is shown using arrows, as depicted in the color wheel. v, center of the vortex[84].
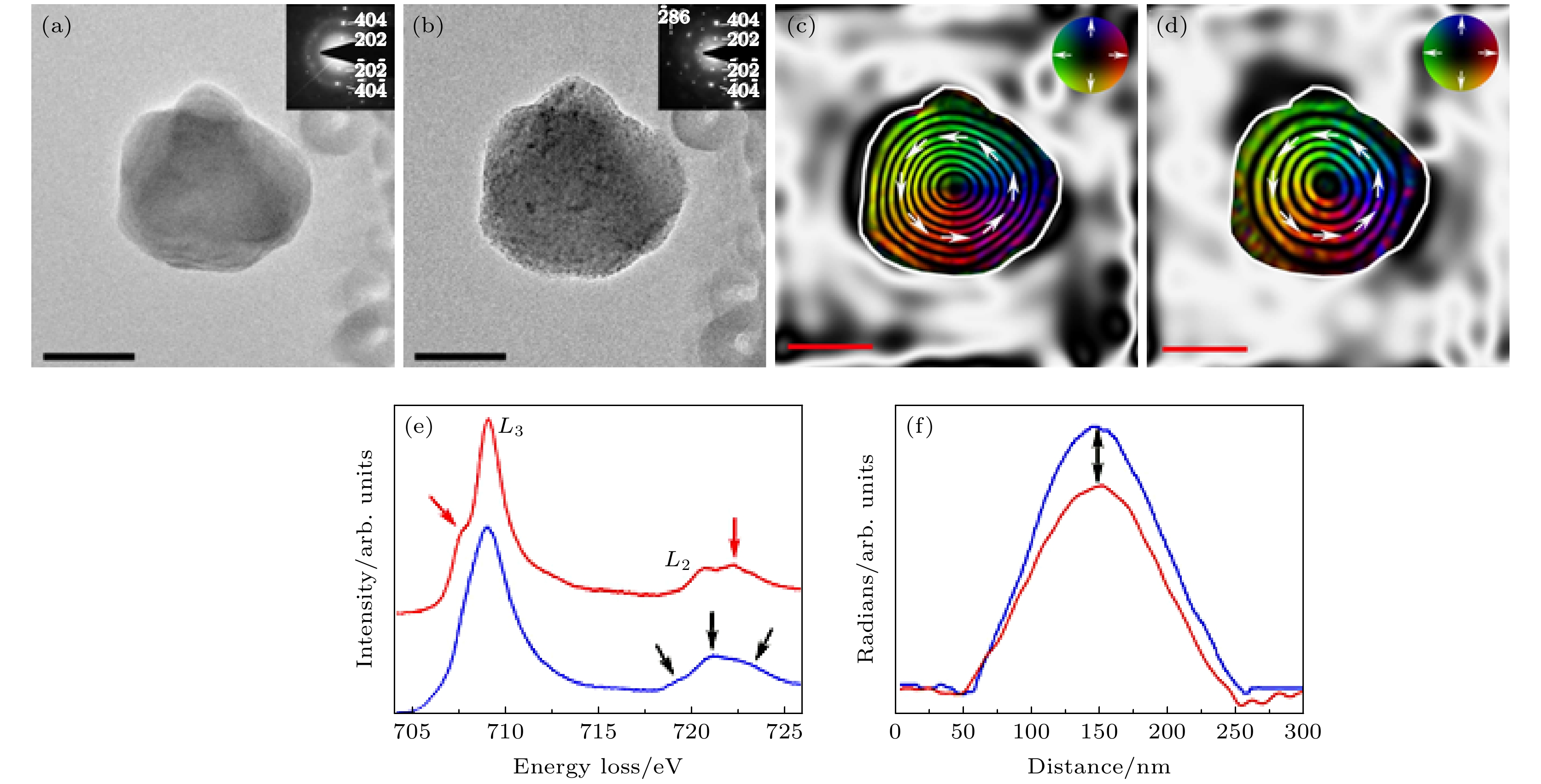
图 9 原位观测氧化态对等轴Fe3O4纳米颗粒磁性的影响, 其中, (a)和(b)图分别是在原位加热至700 ℃ (a)之前和(b)之后的明场透射电子显微像, 原位加热过程是在9 mbar的O2的环境下在环境透射电镜中加热8小时, 插图为对应纳米颗粒的选区电子衍射花样; 从原位加热(c)之前和(d)之后采集的全息图重构出的相位变化信息中获得的磁感应强度分布图, 展示出纳米颗粒的磁涡旋本征态, 等高磁轮廓线的间距是0.79 rad, 磁场方向用箭头表示, 与色轮对应; (e) Fe3O4 颗粒在环境透射电镜中退火前(蓝色, γ-Fe2O3)和退火后(红色, Fe3O4)对应的电子能量损失谱中的Fe 2p的L2, 3边, 黑色箭头表明了Fe3O4 中的三种混合价态, 红色箭头强调了边前峰和边后峰的形成并表明样品在朝着γ-Fe2O3态氧化; (f)退火前(蓝色, γ-Fe2O3)和退火后(红色, Fe3O4) 颗粒中沿颗粒中心至边缘的剩余磁化强度分布, 黑色箭头表明了退火后整体剩磁的减少[85]
Fig. 9. Visualized effect of oxidation on the magnetization of an equiaxed Fe3O4 particle. Bright-field transmission electron microscopy images acquired (a) before and (b) after in situ heating to 700 ℃ under 9 mbar of O2 for 8 h in an environmental transmission electron microscope (ETEM), with associated selected area electron diffraction patterns inset. (c), (d) Magnetic induction maps determined from the magnetic contribution to the phase shift, reconstructed from holograms taken (c) before and (d) after in situ heating, revealing the vortex nature of the particle. The contour spacing is 0.79 rad for both magnetic induction maps. The magnetization direction is shown using arrows, as depicted in the color wheel. (e) Associated electron energy-loss spectra of the Fe 2p L2, 3 edge acquired from the Fe3O4 particle before (blue) and after (red) annealing within the ETEM. Black arrows emphasize three differing intensities from the mixed-valence compound of Fe3O4, while the red arrows highlight formation of pre- and post-peaks that indicate oxidation towards γ-Fe2O3. (f) Line profiles across their centers of magnetic contribution of nanoparticles before (blue) and after (red) annealing. Black arrows in (f) illustrate the loss in overall magnetic remanence[85].
-
[1] Cai R, Martelli F, Vernieres J, Albonetti S, Dimitratos N, Tizaoui C, Palmer R E 2020 ACS Appl. Mater. Interfaces 12 24877
Google Scholar
[2] Curl R F, Smalley R E 1991 Sci. Am. 265 54
[3] Kroto H W, Heath J R, O’Brien S C, Curl R F, Smalley R E 1985 Nature 318 162
Google Scholar
[4] Becker E W, Bier K, Henkes W 1956 Z. Physik 146 333
Google Scholar
[5] Purusottam J, A Welford C Jr 2010 Nanoclusters: a Bridge Across Disciplines (Oxford: Elsevier) pp3−36
[6] Khanna S N, Jena P 1992 Phys. Rev. Lett. 69 1664
Google Scholar
[7] Zhou J, Yang Y, Ercius P, Miao J 2020 MRS Bull. 45 290
Google Scholar
[8] Miller M K 2000 Atom Probe Tomography: Analysis at the Atomic Level (New York: Springer) pp1−23
[9] Midgley P A, Weyland M 2003 Ultramicroscopy 96 413
Google Scholar
[10] Haider M, Uhlemann S, Schwan E, Rose H, Kabius B, Urban K 1998 Nature 392 768
Google Scholar
[11] Midgley P A, Dunin Borkowski R E 2009 Nat. Mater. 8 271
Google Scholar
[12] Urban K W 2009 Nat. Mater. 8 260
Google Scholar
[13] Muller D A 2009 Nat. Mater. 8 263
Google Scholar
[14] Chen F R, Van Dyck D, Kisielowski C 2016 Nat. Commun. 7 10603
Google Scholar
[15] Wiesendanger R, Shvets I V, Bürgler D, Tarrach G, Güntherodt H J, Coey J M, Gräser S 1992 Science 255 583
Google Scholar
[16] Heinze S, Bode M, Kubetzka A, Pietzsch O, Nie X, Blügel S, Wiesendanger R 2000 Science 288 1805
Google Scholar
[17] Kaiser U, Schwarz A, Wiesendanger R 2007 Nature 446 522
Google Scholar
[18] Beaurepaire E, Kappler J P, Krill G, Scheurer F 2010 Magnetism and Synchrotron Radiation (Berlin: Springer) pp145−364
[19] Chao W, Harteneck B, Liddle J, Anderson E, Attwood D 2005 Nature 435 1210
Google Scholar
[20] Kim D H, Fischer P, Chao W, Anderson E, Im M Y, Shin S, Choe S B 2006 J. Appl. Phys. 99 08H303
Google Scholar
[21] Zhu X, Hitchcock A P, Bazylinski D A, Denes P, Joseph J, Lins U, Marchesini S, Shiu H W, Tyliszczak T, Shapiro D A 2016 Proc. Natl. Acad. Sci. U. S. A. 113 E8219
Google Scholar
[22] Pennycook S J 2011 Scanning Transmission Electron Microscopy: Imaging and Analysis (New York: Springer) pp1−90
[23] Jia C, Lentzen M, Urban K 2003 Science 299 870
Google Scholar
[24] Li Z Y, Young N P, Di Vece M, Palomba S, Palmer R E, Bleloch A L, Curley B C, Johnston R L, Jiang J, Yuan J 2008 Nature 451 46
Google Scholar
[25] Bals S, Van Aert S, Romero C P, Lauwaet K, Van Bael M J, Schoeters B, Partoens B, Yücelen E, Lievens P, Van Tendeloo G 2012 Nat. Commun. 3 897
Google Scholar
[26] Lentzen M, Jahnen B, Jia C L, Thust A, Tillmann K, Urban K 2002 Ultramicroscopy 92 233
Google Scholar
[27] Jia C L, Mi S B, Barthel J, Wang D W, Dunin Borkowski R E, Urban K W, Thust A 2014 Nat. Mater. 13 1044
Google Scholar
[28] Van Dyck D, Jinschek J R, Chen F R 2012 Nature 486 243
Google Scholar
[29] Hawkes P W 1980 Computer Processing of Electron Microscope Images (Berlin: Springer) pp1−33
[30] Kirkland E J 1984 Ultramicroscopy 15 151
Google Scholar
[31] Op de Beeck M, Van Dyck D, Coene W 1996 Ultramicroscopy 64 167
Google Scholar
[32] Meyer R R, Kirkland A I, Saxton W O 2002 Ultramicroscopy 92 89
Google Scholar
[33] Allen L J, McBride W, O'Leary N L, Oxley M P 2004 Ultramicroscopy 100 91
Google Scholar
[34] Reich S, Maultzsch J, Thomsen C, Ordejón P 2002 Phys. Rev. B 66 035412
Google Scholar
[35] Miedema M A O, van den Bos A, Buist A H 1994 IEEE Instrum. Meas. Technol. Conf. 43 181
Google Scholar
[36] Buist A H, van den Bos A, Miedema M A O 1996 Ultramicroscopy 64 137
Google Scholar
[37] Iijima S, Ichihashi T 1986 Phys. Rev. Lett. 56 616
Google Scholar
[38] Ajayan P, Marks L 1989 Phys. Rev. Lett. 63 279
Google Scholar
[39] Smith D J, Petford Long A K, Wallenberg L R, Bovin J O 1986 Science 233 872
Google Scholar
[40] Van Dyck D, Lobato I, Chen F R, Kisielowski C 2015 Micron 68 158
Google Scholar
[41] De Rosier D J, Klug A 1968 Nature 217 130
Google Scholar
[42] Hoppe W, Langer R, Knesch G, Poppe C 1968 Naturwissenschaften 55 333
Google Scholar
[43] Hart R G 1968 Science 159 1464
Google Scholar
[44] Arslan I, Yates T J V, Browning N D, Midgley P A 2005 Science 309 2195
Google Scholar
[45] Xia Y, Zhong X Y, Ke X, Zhang G R, Cheng Z, Xu B Q 2016 Small 12 6332
Google Scholar
[46] Zhong X Y, Kabius B, Schreiber D, Eastman J, Fong D, Petford Long A 2012 Appl. Phys. Lett. 100 101604
Google Scholar
[47] Zhong X Y, Kabius B, Schreiber D K, Eastman J A, Fong D D, Petford Long A K 2009 Microsc. microanal. 15 600
Google Scholar
[48] Van Aert S, Batenburg K J, Rossell M D, Erni R, Van Tendeloo G 2011 Nature 470 374
Google Scholar
[49] Miao J W, Ercius P, Billinge S J L 2016 Science 353 aaf2157
Google Scholar
[50] Scott M C, Chen C C, Mecklenburg M, Zhu C, Xu R, Ercius P, Dahmen U, Regan B C, Miao J W 2012 Nature 483 444
Google Scholar
[51] Chen C C, Zhu C, White E R, Chiu C Y, Scott M C, Regan B C, Marks L D, Huang Y, Miao J W 2013 Nature 496 74
Google Scholar
[52] Xu R, Chen C C, Wu L, Scott M C, Theis W, Ophus C, Bartels M, Yang Y, Ramezani Dakhel H, Sawaya M R, Heinz H, Marks L D, Ercius P, Miao J W 2015 Nat. Mater. 14 1099
Google Scholar
[53] Yang Y, Chen C C, Scott M C, Ophus C, Xu R, Pryor A, Wu L, Sun F, Theis W, Zhou J, Eisenbach M, Kent P R C, Sabirianov R F, Zeng H, Ercius P, Miao J 2017 Nature 542 75
Google Scholar
[54] Haruta M, Kobayashi T, Sano H, Yamada N 1987 Chem. Lett. 16 405
Google Scholar
[55] Foster D M, Ferrando R, Palmer R E 2018 Nat. Commun. 9 1323
Google Scholar
[56] Foster D M, Pavloudis T, Kioseoglou J, Palmer R E 2019 Nat. Commun. 10 2583
Google Scholar
[57] Zhou J, Yang Y, Yang Y, Kim D S, Yuan A, Tian X, Ophus C, Sun F, Schmid A K, Nathanson M, Heinz H, An Q, Zeng H, Ercius P, Miao J 2019 Nature 570 500
Google Scholar
[58] Zhu B, Meng J, Yuan W, Zhang X, Yang H, Wang Y, Gao Y 2020 Angew. Chem. Int. Edit. 59 2171
Google Scholar
[59] Altantzis T, Lobato I, De Backer A, Béché A, Zhang Y, Basak S, Porcu M, Xu Q, Sánchez Iglesias A, Liz Marzán L M, Van Tendeloo G, Van Aert S, Bals S 2019 Nano Lett. 19 477
Google Scholar
[60] Abrams I M, McBain J W 1944 J. Appl. Phys. 15 607
Google Scholar
[61] Swift J A, Brown A C 1970 J. Phys. E 3 924
Google Scholar
[62] Zheng H, Smith R, Jun Y W, Kisielowski C, Dahmen U, Alivisatos A 2009 Science 324 1309
Google Scholar
[63] Park J, Zheng H, Lee W C, Geissler P L, Rabani E, Alivisatos A P 2012 ACS Nano 6 2078
Google Scholar
[64] Yuk J M, Park J, Ercius P, Kim K, Hellebusch D, Crommie M, Lee J, Zettl A, Alivisatos A 2012 Science 336 61
Google Scholar
[65] Liao H G, Zherebetskyy D, Xin H, Czarnik C, Ercius P, Elmlund H, Pan M, Wang L W, Zheng H 2014 Science 345 916
Google Scholar
[66] Park J, Elmlund H, Ercius P, Yuk J M, Limmer D T, Chen Q, Kim K, Han S H, Weitz D A, Zettl A, Alivisatos A P 2015 Science 368 290
Google Scholar
[67] Kim B H, Heo J, Kim S, Reboul C F, Chun H, Kang D, Bae H, Hyun H, Lim J, Lee H, Han B, Hyeon T, Alivisatos A P, Ercius P, Elmlund H, Park J 2020 Science 368 60
Google Scholar
[68] Sutter E, Jungjohann K, Bliznakov S, Courty A, Maisonhaute E, Tenney S, Sutter P 2014 Nat. Commun. 5 4946
Google Scholar
[69] Liu S Y, Kundu P, Huang T W, Chuang Y J, Tseng F G, Lu Y, Sui M L, Chen F R 2017 Nano Energy 31 218
Google Scholar
[70] Lu Y, Geng J, Wang K, Zhang W, Ding W, Zhang Z, Xie S, Dai H, Chen F R, Sui M 2017 ACS Nano 11 8018
Google Scholar
[71] Lu Y, Yin W J, Peng K L, Wang K, Hu Q, Selloni A, Chen F R, Liu L M, Sui M L 2018 Nat. Commun. 9 2752
Google Scholar
[72] Schattschneider P, Rubino S, Hébert C, Rusz J, Kuneš J, Novák P, Carlino E, Fabrizioli M, Panaccione G, Rossi G 2006 Nature 441 486
Google Scholar
[73] Wang Z Q, Zhong X Y, Yu R, Cheng Z Y, Zhu J 2013 Nat. Commun. 4 1395
Google Scholar
[74] Rusz J, Eriksson O, Novak P, Oppeneer P 2007 Phys. Rev. B 76 060408(R
Google Scholar
[75] Calmels L, Houdellier F, Warot Fonrose B, Gatel C, Hÿtch M, Serin V, Snoeck E, Schattschneider P 2007 Phys. Rev. B 76 060409(R
Google Scholar
[76] Lin J, Zhong X Y, Rusz J, Kocevski V, Xin H, Cui B, Han L, Lin R, Chen X, Zhu J 2017 Phys. Rev. Mater. 1 071404(R
Google Scholar
[77] Ho P L, Yu C P, Zhang Q, Song K, Buban J P, Choi S Y, Dunin Borkowski R E, Mayer J, Tai N H, Zhu J, Jin L, Zhong X Y 2018 Ultramicroscopy 193 137
Google Scholar
[78] Chen X, Higashikozono S, Ito K, Jin L, Ho P L, Yu C P, Tai N H, Mayer J, Dunin Borkowski R E, Suemasu T, Zhong X Y 2019 Ultramicroscopy 203 37
Google Scholar
[79] Schattschneider P, Stöger Pollach M, Rubino S, Sperl M, Hurm C, Zweck J, Rusz J 2008 Phys. Rev. B 78 104413
Google Scholar
[80] Thersleff T, Rusz J, Hjörvarsson B, Leifer K 2016 Phys. Rev. B 94 134430
Google Scholar
[81] Rusz J, Muto S, Spiegelberg J, Adam R, Tatsumi K, Bürgler D E, Oppeneer P M, Schneider C M 2016 Nat. Commun. 7 12672
Google Scholar
[82] Wang Z, Tavabi A H, Jin L, Rusz J, Tyutyunnikov D, Jiang H, Moritomo Y, Mayer J, Dunin Borkowski R E, Yu R, Zhu J, Zhong X Y 2018 Nat. Mater. 17 221
Google Scholar
[83] 陈鑫峰, 王泽朝, 钟虓䶮 2018 电子显微学报 37 540
Google Scholar
Chen X F, Wang Z C, Zhong X Y 2018 J. Chin. Electrn Microsc. Soc. 37 540
Google Scholar
[84] Almeida T P, Muxworthy A R, Kovács A, Williams W, Brown P D, Dunin Borkowski R E 2016 Sci. Adv. 2 e1501801
Google Scholar
[85] Almeida T P, Kasama T, Muxworthy A R, Williams W, Nagy L, Hansen T W, Brown P D, Dunin Borkowski R E 2014 Nat. Commun. 5 5154
Google Scholar

 下载:
下载:




计量
- 文章访问数: 15189
- PDF下载量: 312
- 被引次数: 0
