










-
钨作为一种重要的核材料, 在辐射环境下的微观演化行为与自间隙原子缺陷的扩散行为密切相关. 研究不同构型自间隙原子的扩散行为有助于全面理解材料的微观演化过程. 本文采用分子动力学方法重点考察了钨中具有不同构型的双自间隙原子随温度变化的扩散行为. 结果表明: 彼此互为最近邻的
$\left\langle 111 \right\rangle$ 双自间隙原子, 随着温度的升高, 从一维扩散演变成三维扩散, 在$ \left\langle 111 \right\rangle$ 方向保持稳定的最近邻结构; 次近邻$ \left\langle 111 \right\rangle$ 双自间隙原子在一定温度范围内沿$ \left\langle 111 \right\rangle$ 方向一维扩散, 当温度高于600 K将解离成两个独立运动的自间隙原子; 而三近邻结构在温度高于300 K就将解离. 非平行结构的双自间隙原子在一定温度范围内形成固着性结构, 几乎不移动, 但在温度高于1000 K时将转化成移动性缺陷. 通过将微动弹性带算法获得的自间隙原子迁移能与阿伦尼乌斯关系拟合的结果进行对比, 表明了钨中单自间隙原子和双自间隙原子的扩散系数随温度的变化规律不适于用阿伦尼乌斯关系来描述, 而线性关系则能合理地描述这一规律.Tungsten, due to its desirable properties (high melting point, low sputtering coefficient, good irradiation resistance etc.), is considered as a promising candidate for the plasma facing materials in future nuclear fusion reactors. Therefore, it will work in extremely harsh environments because it is subjected to the bombadement of high-flux plasma particles and the irradiation of high energy neutrons, resulting in vacancies and interstitials. The migration behavior of self-interstitial atoms is one of the most important factors determining the microstructure evolution in irradiated metals because it will greatly affect the mechanical properties of materials. The study of the diffusion behavior of di-interstitials with different configurations contributes to a better understanding of the self-interstitial atom behavior in tungsten. Despite the inherent difficulty in experimental approaches, atomistic simulation provides an effective means of investigating the defect evolution in materials. In this paper, based on the newly developed interatomic potential for W-W interaction, the diffusion behavior of self-interstitial atoms in tungsten is studied by molecular dynamics simulation. This work focuses on the investigation of the diffusion behavior of di-interstitials with different configurations at different temperatures. The obtained results show that the di-interstitials with the first nearest neighbor configuration presents the one-dimensional migration in the$\left\langle 111 \right\rangle $ direction at temperatures below 1400 K. As the temperature increases, it makes rotations from one$ \left\langle 111 \right\rangle$ - to other$\left\langle 111 \right\rangle $ -directions. Thus migration of di-interstitial atoms with the first nearest neighbor configuration exhibits a change in mechanism from one-dimensional to three-dimensional migration, keeping the stable$\left\langle 111 \right\rangle $ configuration in the whole investigated temperature range. The migration of di-interstitial atoms with the second nearest neighbor configuration is one-dimensional along the$\left\langle 111 \right\rangle$ direction within a certain temperature range. When the temperature is above 600 K, the di-interstitial atoms will dissociate into two individual self-interstitial atoms and move independently. However, the migration of di-interstitial atoms with the third nearest neighbor configuration dissociates at a temperature just above 300 K. The non-parallel self-interstitial atoms form a sessile configuration within a certain temperature range. Once the sessile cluster is formed it can hardly move. Interestingly, it will transform into mobile defect when the temperature is higher than 1000 K. By comparing the migration energy values of these configurations obtained by nudged elastic band method with those of the Arrhenius fits, we find that the diffusivity for each of single- and di-interstitial atoms in tungsten is a linear function of temperature rather than Arrhenius as usually assumed.-
Keywords:
- self-interstitial atoms /
- diffusion behavior /
- irradiation damage /
- molecular dynamics simulation
[1] Chen L, Liu Y L, Zhou H B, Jin S, Zhang Y, Lu G H 2012 Sci. China: Phys. Mech. Astron. 55 614
 Google Scholar
Google Scholar
[2] 汪俊, 周宇璐, 张宝玲, 侯氢 2011 物理学报 60 106601
Google Scholar
Wang J, Zhou Y L, Zhang B L, Hou Q 2011 Acta Phys. Sin. 60 106601
Google Scholar
[3] 郭洪燕, 夏敏, 燕青芝, 郭立平, 陈济红, 葛昌纯 2016 物理学报 65 077803
Google Scholar
Guo H Y, Xia M, Yan Q Z, Guo L P, Chen J H, Ge C C 2016 Acta Phys. Sin. 65 077803
Google Scholar
[4] Zhou W H, Li Y G, Huang L F, Zeng Z, Ju X 2013 J. Nucl. Mater. 437 438
Google Scholar
[5] Zhou W H, Zhang C G, Li Y G, Zeng Z 2014 J. Nucl. Mater. 453 202
Google Scholar
[6] 郭龙婷, 孙继忠, 黄艳, 刘升光, 王德真 2013 物理学报 62 227901
Google Scholar
Guo L T, Sun J Z, Huang Y, Liu S G, Wang D Z 2013 Acta Phys. Sin. 62 227901
Google Scholar
[7] Terentyev D A, Malerba L, Hou M 2007 Phys. Rev. B 75 104108
[8] Marinica M C, Willaime F, Mousseau N 2011 Phys. Rev. B 83 094119
Google Scholar
[9] Fu C C, Willaime F 2004 Phys. Rev. Lett. 92 175503
Google Scholar
[10] Terentyev D A, Klaver T P, Olsson P, Marinica M C, Willaime F, Domain C, Malerba L 2008 Phys Rev Lett. 100 145503
Google Scholar
[11] Derlet P M, Nguyen-Manh D, Dudarev S L 2007 Phys. Rev. B 76 054107
Google Scholar
[12] Ventelon L, Willaime F, Fu C C, Heran M, Ginoux I 2012 J. Nucl. Mater. 425 16
Google Scholar
[13] Tsong T T, Casanova R 1980 Phys. Rev. B 22 4632
Google Scholar
[14] Amino T, Arakawa K, Mori H 2016 Sci. Rep. 6 26099
Google Scholar
[15] Zhou W H, Zhang C G, Li Y G, Zeng Z 2015 Sci. Rep. 4 5096
Google Scholar
[16] Chen D, Hu W, Yang J, Deng H, Sun L, Gao F 2009 Eur. Phys. J. B 68 479
Google Scholar
[17] Dudarev S L, Ma P W 2018 Phys. Rev. Mater. 2 033602
Google Scholar
[18] Ma P W, Dudarev S L 2019 Phys. Rev. Mater. 3 013605
Google Scholar
[19] Swinburne T D, Ma P W, Dudarev S L 2017 New J. Phys. 19 073024
Google Scholar
[20] Bonny G, Terentyev D, Bakaev A, Grigorev P, van Neck D 2014 Modell. Simul. Mater. Sci. Eng. 22 053001
Google Scholar
[21] Marinica M C, Ventelon L, Gilbert M R, Proville L, Dudarev S L, Marian J, Bencteux G, Willaime F 2013 J. Phys.: Condens. Matter 25 395502
Google Scholar
[22] Wang J, Zhou Y L, Li M, Hou Q 2014 Modell. Simul. Mater. Sci. Eng. 22 257
[23] Wang J, Zhou Y L, Li M, Hou Q 2012 J. Nucl. Mater. 427 290
Google Scholar
[24] Wooding S J, Bacon D J, Phythian W J 1995 Philos. Mag. A 72 1261
Google Scholar
[25] Wooding S J, Howe L M, Gao F, Calder A F, Bacon D J 1998 J. Nucl. Mater. 254 191
Google Scholar
[26] Wooding S J, Bacon D J 1997 Philos. Mag. A 76 1033
Google Scholar
[27] Gao F, Bacon D J, Osetsky Y N, Flewitt P E J, Lewis T A 2000 J. Nucl. Mater. 276 213
Google Scholar
[28] Bacon D J, Gao F, Osetsky Yu N 2000 J. Nucl. Mater. 276 1
Google Scholar
[29] Boisvert G, Lewis L J 1996 Phys. Rev. B 54 2880
Google Scholar
[30] Swinburne T D, Dudarev S L 2015 Phys. Rev. B 92 134302
Google Scholar
[31] Jonsson H, Mills G, Jacobsen K W 1998 Classical And Quantum Dynamics In Condensed Phase Simulations (Singapore: World Scientific) pp385−404
-
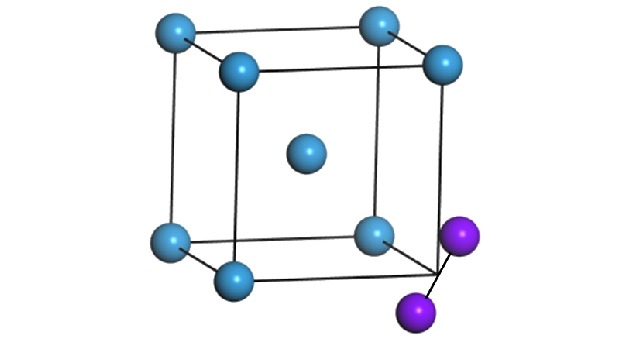
图 1 钨中1-SIA的结构图(紫色球为SIAs结构, 蓝色球为格点原子)
Fig. 1. 1-SIA configuration in W. The purple sphere represents the SIA; the blue one represents the lattice atom.
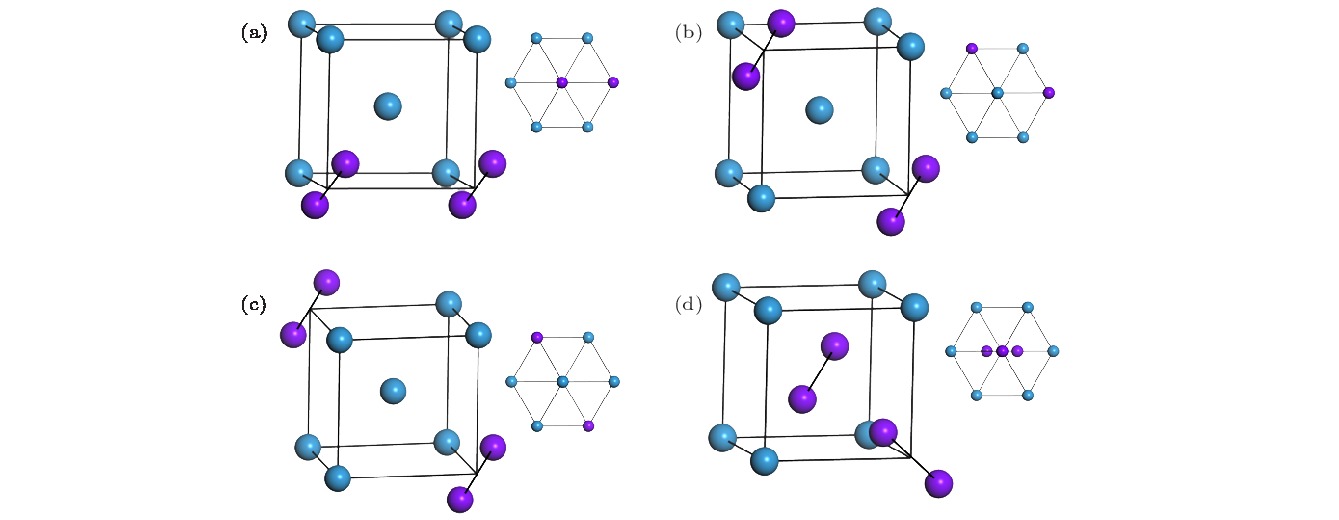
图 2 2-SIAs的不同构型图 (a), (b), (c), (d)分别代表最近邻、次近邻、三近邻以及非平行结构的结构示意图; 右上方的插图分别代表这几种结构
$ \left\langle 111 \right\rangle$ 方向的视图; 紫色球为SIAs, 蓝色球为格点原子Fig. 2. Schematic illustrations of the 2-SIAs with different configurations: (a), (b), (c), (d) Represent the configuration of the 2-SIAs-1st, 2-SIAs-2nd, 2-SIAs-3rd and the non-parallel SIAs, respectively. Insets represent the views corresponding to their
$ \left\langle 111 \right\rangle$ orientations; the purple sphere stands for the SIA and the blue one stands for the lattice atom.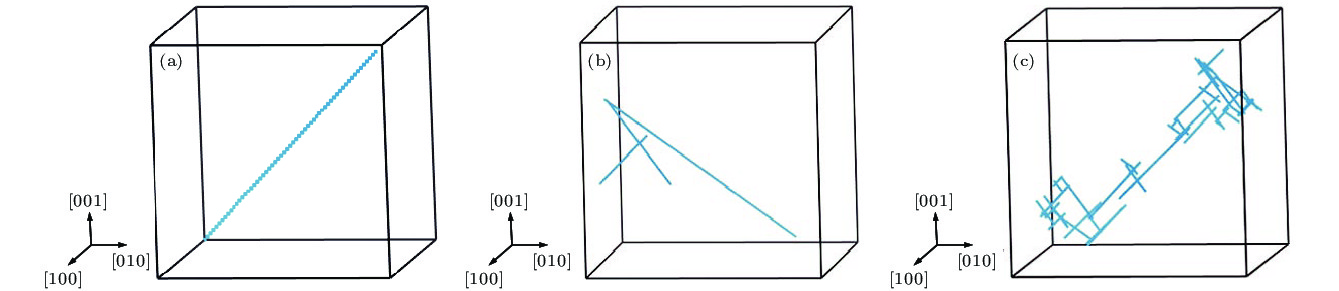
图 3 1-SIAs在不同温度下演化10 ns的扩散径迹图 (a) T = 100 K; (b) T = 700 K; (c) T = 1000 K
Fig. 3. Diffusive trajectories of 1-SIA for temperatures of (a) 100 K, (b) 700 K and (c) 1000 K.
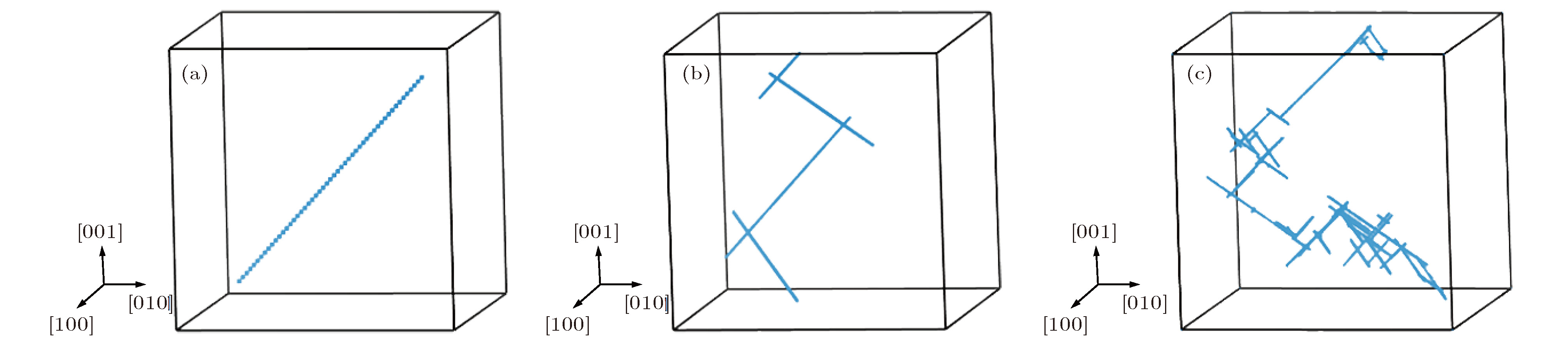
图 4 最近邻结构在不同温度下演化10 ns的扩散径迹图 (a) T = 100 K; (b) T = 1400 K; (c) T = 2000 K
Fig. 4. Diffusive trajectories of 2-SIAs-1st for temperatures of (a) 100 K, (b) 1400 K and (c) 2000 K.
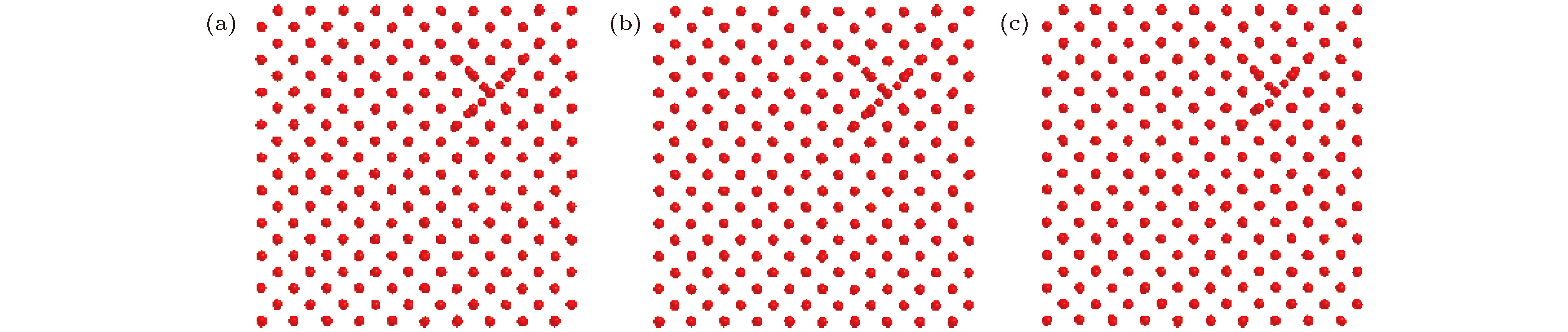
图 5 T = 500 K时sessile结构在不同时间的结构图 (a) t = 0.5 ns; (b) t = 2 ns; (c) t = 5 ns
Fig. 5. Views of the sessile cluster obtained by molecular dynamics simulation at different time when T = 500 K: (a) t = 0.5 ns; (b) t = 2 ns; (c) t = 5 ns.
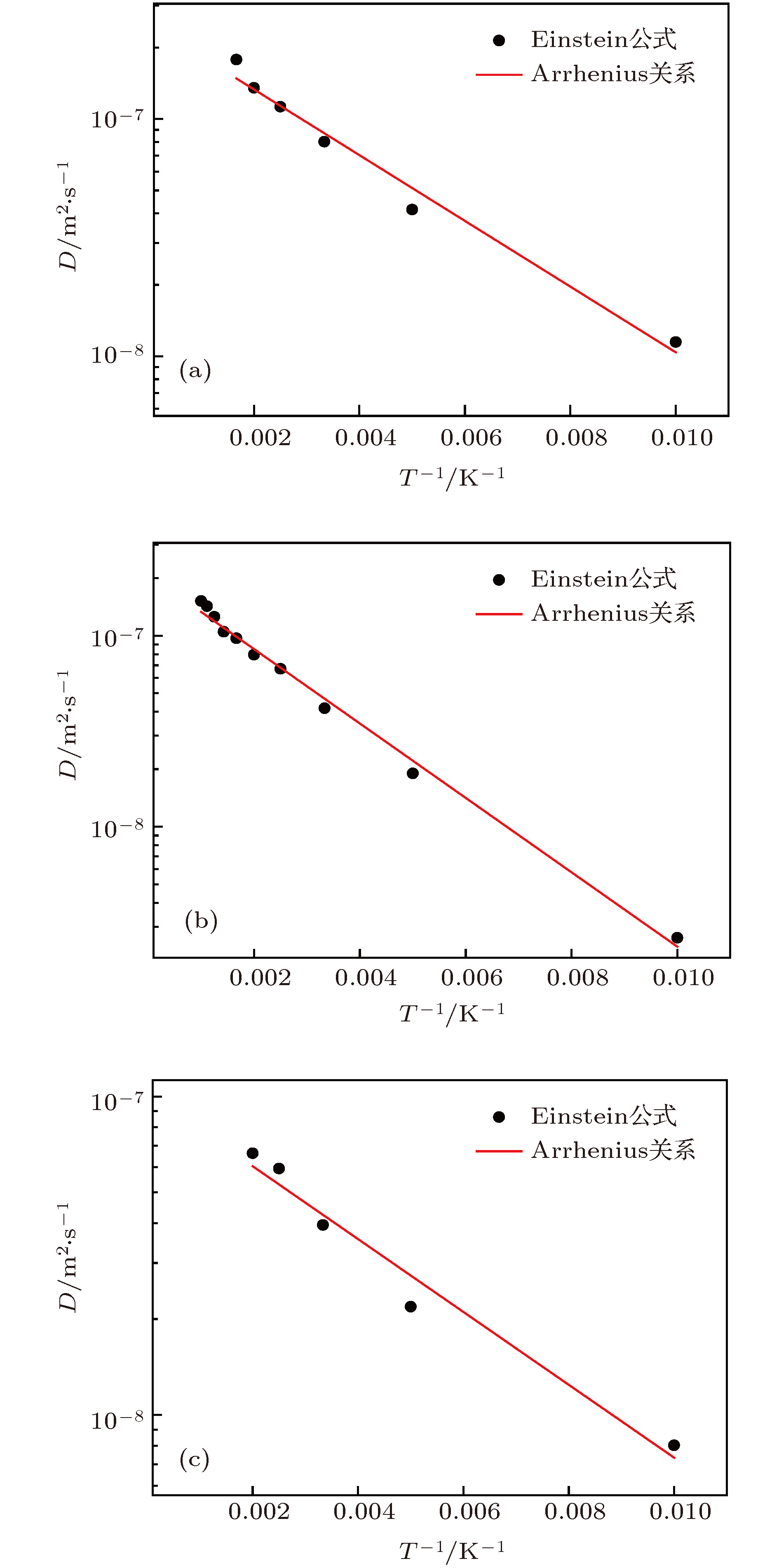
图 6 不同结构的扩散系数(图中实线是根据Arrhenius关系拟合的结果) (a) 1-SIA; (b) 2-SIAs-1st; (c) 2-SIAs-2nd
Fig. 6. Arrhenius plots of diffusion coefficients of single SIA and di-interstitial atoms in tungsten, which is determined using MD simulations and plotted as a function of the absolute temperature T: (a) 1-SIA; (b) 2-SIAs-1st; (c) 2-SIAs-2nd.
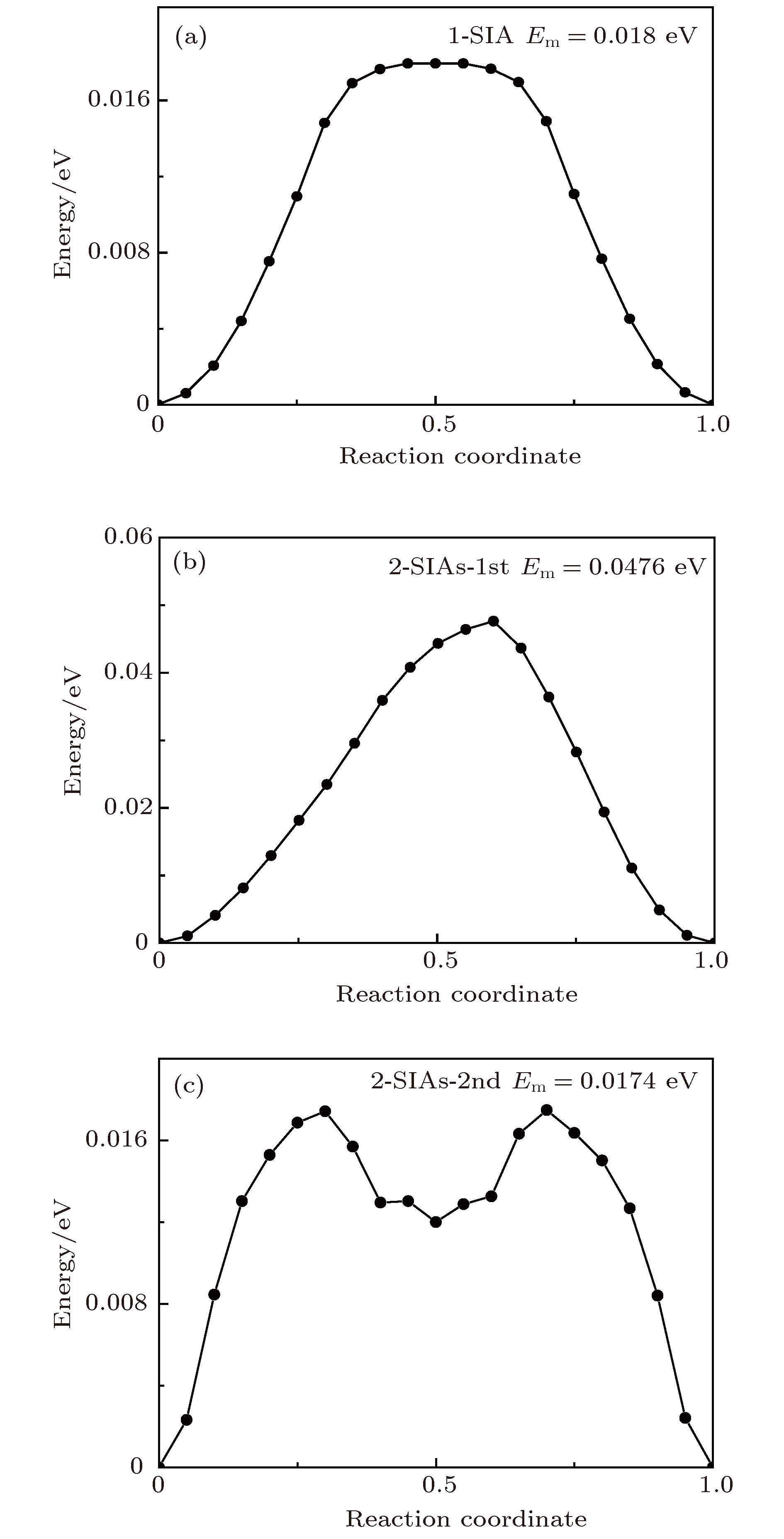
图 7 通过NEB方法所得不同结构的迁移能垒 (a) 1-SIA; (b) 2-SIAs-1st; (c) 2-SIAs-2nd
Fig. 7. Migration barriers for SIAs with different structures studied by NEB method: (a) 1-SIA; (b) 2-SIAs-1st; (c) 2-SIAs-2nd.
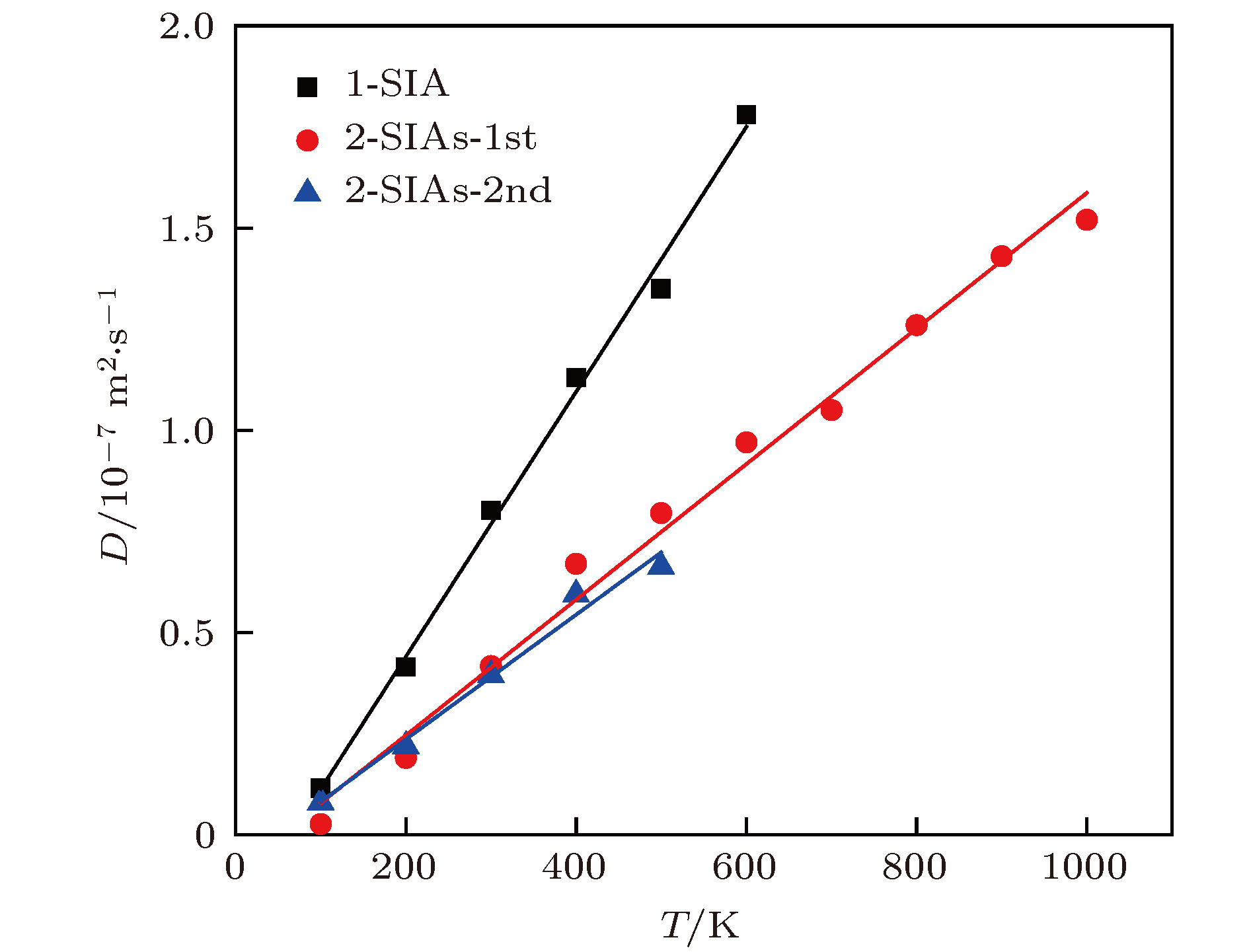
图 8 不同缺陷的扩散系数
Fig. 8. Diffusion coefficient for self-interstitials of different configuration in tungsten determined by molecular dynamics simulations and plotted as a function of the absolute temperature T (the solid lines are linear fits).
表 1 不同缺陷结构的形成能
Table 1. Formation energies of self-interstitials with different configurations.
缺陷类型 能量/eV 1SIA 10.406 2-SIAs-1st 18.574 2-SIAs-2nd 20.510 2-SIAs-3rd 20.808 sessile cluster 19.330  下载: 导出CSV
下载: 导出CSV
表 2 不同缺陷结构的束缚能
Table 2. Binding energies of self-interstitials with different configurations.
缺陷类型 能量/eV 2-SIAs-1st 2.245 2-SIAs-2nd 0.309 2-SIAs-3rd 0.012 sessile cluster 1.489
下载: 导出CSV
表 3 Arrhenius拟合所得各个间隙结构的扩散迁移能与前因子
Table 3. Migration energy Em (in eV) and prefactor D0 (in cm2/s) for W clusters diffusion obtained by Arrhenius fitting.
缺陷结构 迁移能${E_{\rm{m}}}/{\rm{eV}}$(Arrhenius) 前因子D0/m2·s–1 迁移能${E_{\rm{m}}}/{\rm{eV}}$ (NEB) 1-SIA 0.0274 2.52 × 10–7 0.0180 2-SIAs-1st 0.0386 2.08 × 10–7 0.0476 2-SIAs-2nd 0.0248 1.02 × 10–7 0.0174
下载: 导出CSV
-
[1] Chen L, Liu Y L, Zhou H B, Jin S, Zhang Y, Lu G H 2012 Sci. China: Phys. Mech. Astron. 55 614
Google Scholar
[2] 汪俊, 周宇璐, 张宝玲, 侯氢 2011 物理学报 60 106601
Google Scholar
Wang J, Zhou Y L, Zhang B L, Hou Q 2011 Acta Phys. Sin. 60 106601
Google Scholar
[3] 郭洪燕, 夏敏, 燕青芝, 郭立平, 陈济红, 葛昌纯 2016 物理学报 65 077803
Google Scholar
Guo H Y, Xia M, Yan Q Z, Guo L P, Chen J H, Ge C C 2016 Acta Phys. Sin. 65 077803
Google Scholar
[4] Zhou W H, Li Y G, Huang L F, Zeng Z, Ju X 2013 J. Nucl. Mater. 437 438
Google Scholar
[5] Zhou W H, Zhang C G, Li Y G, Zeng Z 2014 J. Nucl. Mater. 453 202
Google Scholar
[6] 郭龙婷, 孙继忠, 黄艳, 刘升光, 王德真 2013 物理学报 62 227901
Google Scholar
Guo L T, Sun J Z, Huang Y, Liu S G, Wang D Z 2013 Acta Phys. Sin. 62 227901
Google Scholar
[7] Terentyev D A, Malerba L, Hou M 2007 Phys. Rev. B 75 104108
[8] Marinica M C, Willaime F, Mousseau N 2011 Phys. Rev. B 83 094119
Google Scholar
[9] Fu C C, Willaime F 2004 Phys. Rev. Lett. 92 175503
Google Scholar
[10] Terentyev D A, Klaver T P, Olsson P, Marinica M C, Willaime F, Domain C, Malerba L 2008 Phys Rev Lett. 100 145503
Google Scholar
[11] Derlet P M, Nguyen-Manh D, Dudarev S L 2007 Phys. Rev. B 76 054107
Google Scholar
[12] Ventelon L, Willaime F, Fu C C, Heran M, Ginoux I 2012 J. Nucl. Mater. 425 16
Google Scholar
[13] Tsong T T, Casanova R 1980 Phys. Rev. B 22 4632
Google Scholar
[14] Amino T, Arakawa K, Mori H 2016 Sci. Rep. 6 26099
Google Scholar
[15] Zhou W H, Zhang C G, Li Y G, Zeng Z 2015 Sci. Rep. 4 5096
Google Scholar
[16] Chen D, Hu W, Yang J, Deng H, Sun L, Gao F 2009 Eur. Phys. J. B 68 479
Google Scholar
[17] Dudarev S L, Ma P W 2018 Phys. Rev. Mater. 2 033602
Google Scholar
[18] Ma P W, Dudarev S L 2019 Phys. Rev. Mater. 3 013605
Google Scholar
[19] Swinburne T D, Ma P W, Dudarev S L 2017 New J. Phys. 19 073024
Google Scholar
[20] Bonny G, Terentyev D, Bakaev A, Grigorev P, van Neck D 2014 Modell. Simul. Mater. Sci. Eng. 22 053001
Google Scholar
[21] Marinica M C, Ventelon L, Gilbert M R, Proville L, Dudarev S L, Marian J, Bencteux G, Willaime F 2013 J. Phys.: Condens. Matter 25 395502
Google Scholar
[22] Wang J, Zhou Y L, Li M, Hou Q 2014 Modell. Simul. Mater. Sci. Eng. 22 257
[23] Wang J, Zhou Y L, Li M, Hou Q 2012 J. Nucl. Mater. 427 290
Google Scholar
[24] Wooding S J, Bacon D J, Phythian W J 1995 Philos. Mag. A 72 1261
Google Scholar
[25] Wooding S J, Howe L M, Gao F, Calder A F, Bacon D J 1998 J. Nucl. Mater. 254 191
Google Scholar
[26] Wooding S J, Bacon D J 1997 Philos. Mag. A 76 1033
Google Scholar
[27] Gao F, Bacon D J, Osetsky Y N, Flewitt P E J, Lewis T A 2000 J. Nucl. Mater. 276 213
Google Scholar
[28] Bacon D J, Gao F, Osetsky Yu N 2000 J. Nucl. Mater. 276 1
Google Scholar
[29] Boisvert G, Lewis L J 1996 Phys. Rev. B 54 2880
Google Scholar
[30] Swinburne T D, Dudarev S L 2015 Phys. Rev. B 92 134302
Google Scholar
[31] Jonsson H, Mills G, Jacobsen K W 1998 Classical And Quantum Dynamics In Condensed Phase Simulations (Singapore: World Scientific) pp385−404
-
[1] 韦昭召. 不同取向B2结构FeAl合金纳米线弯曲行为的分子动力学模拟. 物理学报, 2025, 74(3): 036201. doi: 10.7498/aps.74.20241030 [2] 曹嵩, 殷雯, 周斌, 胡志良, 沈飞, 易天成, 王松林, 梁天骄. 中国散裂中子源二期靶站关键部件辐照损伤模拟计算. 物理学报, 2024, 73(9): 092501. doi: 10.7498/aps.73.20240088 [3] 秦梦飞, 王英敏, 张红玉, 孙继忠. 〈100〉 间隙型位错环在纯钨及含氦杂质钨(010)表面下运动行为的分子动力学模拟. 物理学报, 2023, 72(24): 245204. doi: 10.7498/aps.72.20230651[4] 明知非, 宋海洋, 安敏荣. 基于分子动力学模拟的石墨烯镁基复合材料力学行为. 物理学报, 2022, 71(8): 086201. doi: 10.7498/aps.71.20211753 [5] 魏雯静, 高旭东, 吕亮亮, 许楠楠, 李公平. 中子对碲锌镉辐照损伤模拟研究. 物理学报, 2022, 71(22): 226102. doi: 10.7498/aps.71.20221195 [6] 潘伶, 张昊, 林国斌. 纳米液滴撞击柱状固体表面动态行为的分子动力学模拟. 物理学报, 2021, 70(13): 134704. doi: 10.7498/aps.70.20210094 [7] 周边, 杨亮. 分子动力学模拟冷却速率对非晶合金结构与变形行为的影响. 物理学报, 2020, 69(11): 116101. doi: 10.7498/aps.69.20191781 [8] 韦昭召, 马骁, 柯常波, 张新平. Fe合金FCC-BCC原子尺度台阶型马氏体相界面迁移行为的分子动力学模拟研究. 物理学报, 2020, 69(13): 136102. doi: 10.7498/aps.69.20191903 [9] 梁晋洁, 高宁, 李玉红. 体心立方Fe中 ${ \langle 100 \rangle}$ 位错环对微裂纹扩展影响的分子动力学研究. 物理学报, 2020, 69(11): 116102. doi: 10.7498/aps.69.20200317[10] 梁晋洁, 高宁, 李玉红. 表面效应对铁 ${\left\langle 100 \right\rangle} $ 间隙型位错环的影响. 物理学报, 2020, 69(3): 036101. doi: 10.7498/aps.69.20191379[11] 崔振国, 勾成俊, 侯氢, 毛莉, 周晓松. 低能中子在锆中产生的辐照损伤的计算机模拟研究. 物理学报, 2013, 62(15): 156105. doi: 10.7498/aps.62.156105 [12] 夏冬, 王新强. 超细Pt纳米线结构和熔化行为的分子动力学模拟研究. 物理学报, 2012, 61(13): 130510. doi: 10.7498/aps.61.130510 [13] 朱勇, 李宝华, 谢国锋. 质子对BaTiO3薄膜辐照损伤的计算机模拟. 物理学报, 2012, 61(4): 046103. doi: 10.7498/aps.61.046103 [14] 贺平逆, 宁建平, 秦尤敏, 赵成利, 苟富均. 低能Cl原子刻蚀Si(100)表面的分子动力学模拟. 物理学报, 2011, 60(4): 045209. doi: 10.7498/aps.60.045209 [15] 汪俊, 张宝玲, 周宇璐, 侯氢. 金属钨中氦行为的分子动力学模拟. 物理学报, 2011, 60(10): 106601. doi: 10.7498/aps.60.106601 [16] 颜超, 段军红, 何兴道. 低能原子沉积在Pt(111)表面的分子动力学模拟. 物理学报, 2010, 59(12): 8807-8813. doi: 10.7498/aps.59.8807 [17] 贺新福, 杨文, 樊胜. 论FeCr合金辐照损伤的多尺度模拟. 物理学报, 2009, 58(12): 8657-8669. doi: 10.7498/aps.58.8657 [18] 孟丽娟, 李融武, 刘绍军, 孙俊东. 异质原子在Cu(001)表面扩散的分子动力学模拟. 物理学报, 2009, 58(4): 2637-2643. doi: 10.7498/aps.58.2637 [19] 葛四平, 朱 星, 杨威生. 用扫描隧道显微镜操纵Cu亚表面自间隙原子. 物理学报, 2005, 54(2): 824-831. doi: 10.7498/aps.54.824 [20] 唐 鑫, 张 超, 张庆瑜. Cu(111)三维表面岛对表面原子扩散影响的分子动力学研究. 物理学报, 2005, 54(12): 5797-5803. doi: 10.7498/aps.54.5797






 下载:
下载:





计量
- 文章访问数: 19153
- PDF下载量: 138
- 被引次数: 0
