










-
表面等离激元是金属与介质表面自由电子的集体振荡, 能够突破衍射极限, 将光束缚在纳米结构表面附近极小的空间内, 为纳米尺度的光操控提供可能. 利用表面等离激元共振, 不仅可以增强局域表面电磁场强度, 实现对表面附近分子荧光和拉曼信号的极大增强, 而且等离激元弛豫诱导的热电子还可以调控表面分子的化学反应, 提高反应速率和选择性, 即等离激元调控(催化)化学反应. 作为一种新型催化体系, 等离激元催化已经实现了多种传统光催化中难以发生的化学反应, 是表面等离激元领域的前沿热点问题. 由于等离激元催化反应的复杂性与多样性, 其反应动力学过程的完全表征和反应机理的揭示仍然是一个巨大的挑战. 精确表征催化反应的中间及最终产物, 获取反应动力学过程中更多的细节信息, 对于探索等离激元催化机理, 以及设计更为合理高效的催化体系极为重要. 本文围绕等离激元催化的最新研究进展, 总结并探讨等离激元催化中所使用的各种表征技术. 首先, 简单介绍了等离激元催化的基本概念和催化机理. 其次, 综述了拉曼光谱(包括表面/针尖增强拉曼光谱), 在等离激元催化原位监测中的应用, 并进一步详细介绍了气相色谱法、气相色谱-质谱联用、高效液相色谱法、扫描透射电子显微镜、扫描隧道显微镜、扫描电化学显微镜、紫外可见吸收光谱等技术在等离激元催化反应研究中的重要作用. 最后, 探讨了这些表征技术在等离激元催化动力学过程研究和催化机理探索中的特点与优势, 并展望了等离激元催化及相关表征技术的发展与挑战.Surface plasmons are collective oscillations of free electrons at the interface between metal and dielectric. Surface plasmons can break through the diffraction limit of light, because the electromagnetic field is confined in a very small space near the surface of the nanostructure, which provides a possibility for nanometer-scale light manipulation. By using surface plasmon resonance, the local surface electromagnetic field can be strongly enhanced, which can be used to enhance the molecular fluorescence and Raman signals. In addition, the plasmon relaxation induces thermal electrons which can drive the catalytic reaction of surface molecules to achieve a selective catalytic reaction at normal temperature, which is so-called plasmon mediated chemical reaction (or plasmonic catalysis). As a new type of catalytic system, plasmonic catalysis can mediate chemical reactions that are difficult to occur under various conventional conditions. Due to the complexity and diversity of plasmon catalyzed reactions, it is still a huge challenge to fully characterize the reaction kinetics and understand its reaction mechanism. Characterizing the intermediate and final products in the catalytic reaction accurately and obtaining more detailed information in the reaction process are essential for exploring the theoretical mechanism of plasmon catalysis. In this paper, we review the characterization techniques used in plasmon catalysis in detail in the progress of plasmon catalysis. First, the basic concepts of plasmon catalysis and several common catalytic mechanisms are introduced. Second, the Raman spectroscopy, including the application of surface and tip-enhanced Raman spectroscopy in plasmon catalytic in situ monitoring are reviewed. Then, the other techniques such as gas chromatography, gas chromatography-mass spectrometry, high performance liquid chromatography, scanning transmission electron microscopy, scanning tunneling microscopy, scanning electrochemical microscopy and UV-visible absorption spectroscopy for monitoring plasmon catalyzed reaction are introduced in detail. Finally, the characteristics and advantages of these characterization techniques in the study of kinetic catalytic process and catalytic mechanism of plasmon, and the future development and challenge are mentioned and analyzed.
-
Keywords:
- surface plasmon resonance /
- plasmonic catalysis /
- characterization
[1] Zhang Z, Deckert G T, Deckert V 2015 Analyst 140 4325
 Google Scholar
Google Scholar
[2] Jiang R, Zhang M, Qian S L, Yan F, Pei L Q, Jin S, Zhao L B, Wu D Y, Tian Z Q 2016 J. Phys. Chem. C 120 16427
Google Scholar
[3] Tesema T E, Kafle B, Tadesse M G, Habteyes T G 2017 J. Phys. Chem. C 121 7421
[4] Zhang X, Li X, Zhang D, Su N Q, Yang W, Everitt H O, Liu J 2017 Nat. Commun. 8 14542
Google Scholar
[5] Zhou L, Swearer D F, Zhang C, Robatjazi H, Zhao H, Henderson L, Dong L, Christopher P, Carter E A, Nordlander P, Halas N J 2018 Science 362 69
Google Scholar
[6] Kazuma E, Jung J, Ueba H, Trenary M, Kim Y 2018 Science 360 521
Google Scholar
[7] Zhan C, Chen X J, Yi J, Li J F, Wu D Y, Tian Z Q 2018 Nat. Rev. Chem. 2 216
Google Scholar
[8] Liu Z, Hou W, Pavaskar P, Aykol M, Cronin S B 2011 Nano Lett. 11 1111
Google Scholar
[9] Chen J, Bailey C S, Hong Y, Wang L, Cai Z, Shen L, Hou B, Wang Y, Shi H, Sambur J, Ren W, Pop E, Cronin S B 2019 ACS Photonics 6 787
Google Scholar
[10] Kazuma E, Kim Y 2019 Angew. Chem. Int. Ed. 58 4800
Google Scholar
[11] Xie W, Schlücker S 2015 Nat. Commun. 6 7570
Google Scholar
[12] Zhang Y, Nelson T, Tretiak S, Guo H, Schatz G C 2018 ACS Nano 12 8415
Google Scholar
[13] Wu K, Chen J, McBride J R, Lian T 2015 Science 349 632
Google Scholar
[14] Duchene J S, Tagliabue G, Welch A J, Cheng W H, Atwater H A 2018 Nano Lett. 18 2545
Google Scholar
[15] Golubev A A, Khlebtsov B N, Rodriguez R D, Chen Y, Zahn D R T 2018 J. Phys. Chem. C 122 5657
[16] Zhang X, Li X, Reish M E, Zhang D, Su N Q, Gutiérrez Y, Moreno F, Yang W, Everitt H O, Liu J 2018 Nano Lett. 18 1714
Google Scholar
[17] Jeanmaire D L, Van R P 1977 J. Electroanal. Chem. Interfacial Electrochem. 84 1
Google Scholar
[18] Albrecht M G, Creighton J A 1977 J. Am. Chem. Soc. 99 5215
Google Scholar
[19] Kleinman S L, Frontiera R R, Henry A I, Dieringer J A, Duyne R P V 2012 Phys. Chem. Chem. Phys. 15 21
[20] Zhang C, Jiang S Z, Huo Y Y, Liu A H, Xu S C, Liu X Y, Sun Z C, Xu Y Y, Li Z, Man B Y 2015 Opt. Express 23 24811
Google Scholar
[21] Cao E, Lin W, Sun M, Liang W, Song Y 2018 Nanophotonics 7 145
Google Scholar
[22] Li C, Yu J, Xu S, Jiang S, Xiu X, Chen C, Liu A, Wu T, Man B, Zhang C 2018 Adv. Mater. Technol. 3 1800174
Google Scholar
[23] Xu J, Li C, Si H, Zhao X, Wang L, Jiang S, Wei D, Yu J, Xiu X, Zhang C 2018 Opt. Express 26 21546
Google Scholar
[24] Zhang C, Li C, Yu J, Jiang S, Xu S, Yang C, Liu Y J, Gao X, Liu A, Man B 2018 Sens. Actuators B: Chem. 258 163
Google Scholar
[25] Zhang Z, Sheng S, Wang R, Sun M 2016 Anal. Chem. 88 9328
Google Scholar
[26] Anderson M S 2000 Appl. Phys. Lett. 76 3130
Google Scholar
[27] Hayazawa N, Inouye Y, Sekkat Z, Kawata S 2000 Opt. Commun. 183 333
Google Scholar
[28] Stöckle R M, Suh Y D, Deckert V, Zenobi R 2000 Chem. Phys. Lett. 318 131
Google Scholar
[29] Verma P 2017 Chem. Rev. 117 6447
Google Scholar
[30] Han S W, Lee I, Kim K 2002 Langmuir 18 182
Google Scholar
[31] Huang Y F, Zhu H P, Liu G K, Wu D Y, Ren B, Tian Z Q 2010 J. Am. Chem. Soc. 132 9244
Google Scholar
[32] Fang Y, Li Y, Xu H, Sun M 2010 Langmuir 26 7737
Google Scholar
[33] Dong B, Fang Y, Chen X, Xu H, Sun M 2011 Langmuir 27 10677
Google Scholar
[34] Zhang Z, Merk V, Hermanns A, Unger W E S, Kneipp J 2017 ACS Catal. 7 7803
Google Scholar
[35] Zhang Z, Kinzel D, Deckert V 2016 J. Phys. Chem. C 120 20978
Google Scholar
[36] Li J F, Huang Y F, Ding Y, Yang Z L, Li S B, Zhou X S, Fan F R, Zhang W, Zhou Z Y, Wu D Y, Ren B, Wang Z L, Tian Z Q 2010 Nature 464 392
Google Scholar
[37] Wang Y H, Wei J, Radjenovic P, Tian Z Q, Li J F 2019 Anal. Chem. 91 1675
Google Scholar
[38] Xie W, Walkenfort B, Schlücker S 2013 J. Am. Chem. Soc. 135 1657
Google Scholar
[39] Zhang H, Wang C, Sun H L, Fu G, Chen S, Zhang Y J, Chen B H, Anema J R, Yang Z L, Li J F, Tian Z Q 2017 Nat. Commun. 8 15447
Google Scholar
[40] Kneipp K, Wang Y, Kneipp H, Perelman L T, Itzkan I, Dasari R R, Feld M S 1997 Phys. Rev. Lett. 78 1667
Google Scholar
[41] Nie S, Emory S R 1997 Science 275 1102
Google Scholar
[42] Chen H Y, Lin M H, Wang C Y, Chang Y M, Gwo S 2015 J. Am. Chem. Soc. 137 13698
Google Scholar
[43] Marshall A R L, Stokes J, Viscomi F N, Proctor J E, Gierschner J, Bouillard J S G, Adawi A M 2017 Nanoscale 9 17415
Google Scholar
[44] Kim N H, Hwang W, Baek K, Rohman M R, Kim J, Kim H W, Mun J, Lee S Y, Yun G, Murray J, Ha J W, Rho J, Moskovits M, Kim K 2018 J. Am. Chem. Soc. 140 4705
Google Scholar
[45] Santos D P, Temperini M L A, Brolo A G 2019 Acc. Chem. Res. 52 456
Google Scholar
[46] Zhang Z, Deckert G T, Singh P, Deckert V 2015 Chem. Commun. 51 3069
Google Scholar
[47] Choi H K, Park W H, Park C G, Shin H H, Lee K S, Kim Z H 2016 J. Am. Chem. Soc. 138 4673
Google Scholar
[48] Sprague E A, McAnally M O, Zhdanov D V, Zrimsek A B, Apkarian V A, Seideman T, Schatz G C, van Duyne R P 2017 J. Am. Chem. Soc. 139 15212
Google Scholar
[49] Brooks J L, Frontiera R R 2016 J. Phys. Chem. C 120 20869
Google Scholar
[50] Brandt N C, Keller E L, Frontiera R R 2016 J. Phys. Chem. Lett. 7 3179
Google Scholar
[51] Keller E L, Frontiera R R 2018 ACS Nano 12 5848
Google Scholar
[52] Schrojenstein E M, Deckert G T, Mank A J G, Deckert V, Weckhuysen B M 2012 Nat. Nanotechnol. 7 583
Google Scholar
[53] Sun M, Zhang Z, Zheng H, Xu H 2012 Sci. Rep. 2 647
Google Scholar
[54] Zhong J H, Jin X, Meng L, Wang X, Su H S, Yang Z L, Williams C T, Ren B 2017 Nat. Nanotechnol. 12 132
Google Scholar
[55] Su H S, Zhang X G, Sun J J, Jin X, Wu D Y, Lian X B, Zhong J H, Ren B 2018 Angew. Chem. Int. Ed. 57 13177
Google Scholar
[56] Zhang Z, Sheng S, Zheng H, Xu H, Sun M 2014 Nanoscale 6 4903
Google Scholar
[57] Zhang Z, Chen L, Sun M, Ruan P, Zheng H, Xu H 2013 Nanoscale 5 3249
Google Scholar
[58] Zhang Z, Richard L M, Deckert V 2017 Faraday Discuss. 205 213
Google Scholar
[59] Yu S, Wilson A J, Heo J, Jain P K 2018 Nano Lett. 18 2189
Google Scholar
[60] Hallett G L, Silvero M J, González M, Grenier M, Netto J C, Scaiano J C 2011 J. Phys. Chem. C 115 10784
Google Scholar
[61] Vadai M, Angell D K, Hayee F, Sytwu K, Dionne J A 2018 Nat. Commun. 9 4658
Google Scholar
[62] Böckmann H, Gawinkowski S, Waluk J, Raschke M B, Wolf M, Kumagai T 2018 Nano Lett. 18 152
Google Scholar
[63] Yu Y, Sundaresan V, Willets K A 2018 J. Phys. Chem. C 122 5040
Google Scholar
[64] Chen K H, Pu Y C, Chang K D, Liang Y F, Liu C M, Yeh J W, Shih H C, Hsu Y J 2012 J. Phys. Chem. C 116 19039
Google Scholar
-
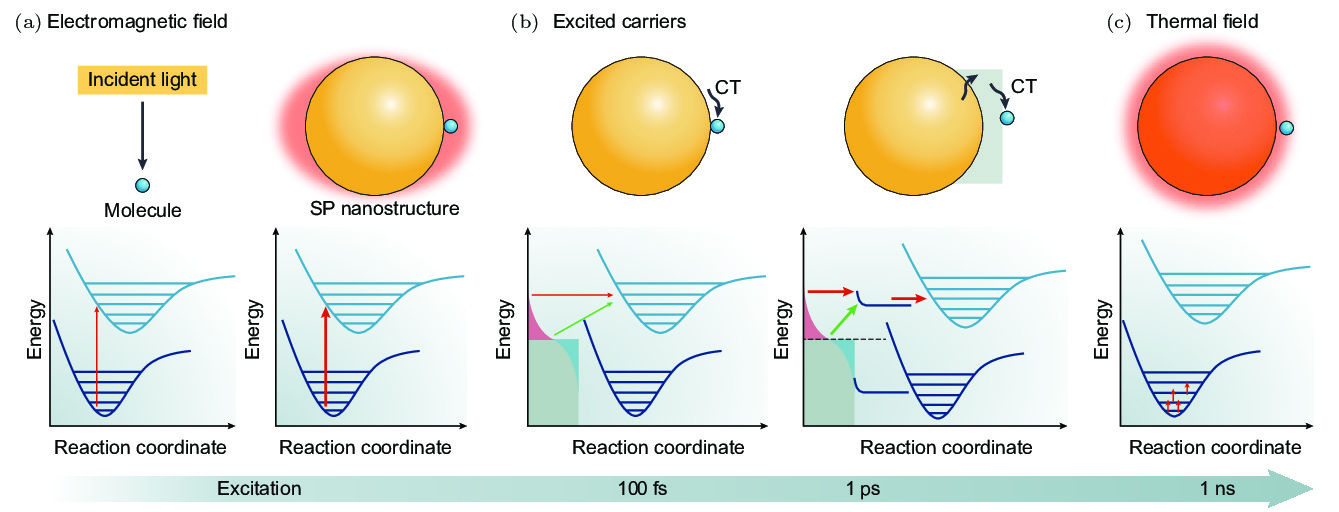
图 1 等离激元调控化学反应机理示意图[7] (a) 等离激元产生的局域电磁场增强导致分子发生反应的可能性增加, 从而提高分子化学反应的速率或产率; (b) 等离激元弛豫产生的热电子通过直接或者间接的方式转移给吸附在金属表面的分子, 导致分子发生化学反应; (c) 颗粒的局域温度升高从而加快化学反应速率
Fig. 1. Schematic diagram of the mechanism of plasmon mediated chemical reaction[7]: (a) The enhanced localized electromagnetic field generated by plasmon increases the probability of molecules reaction, thereby increasing the rate or yield of chemical reaction; (b) the hot electrons generated by the plasmon relaxation are directly or indirectly transferred to the adsorbed molecules, resulting in chemical reaction of molecules; (c) with the increase of the local temperature of the particles, the chemical reaction accelerates.

图 2 (a) 通过SERS光谱检测的Ag基底上PATP分子到DMAB分子的转变[31]; (b) 在Ag纳米颗粒上PATP分子反应生成DMAB分子的时间依赖SERS光谱[32]
Fig. 2. (a) Conversion of PATP molecules to DMAB molecules on Ag substrate and the SERS spectroscopy[31]; (b) time-dependent SERS spectroscopy of DMAB molecules generated by PATP molecule reaction on Ag nanoparticles[32].
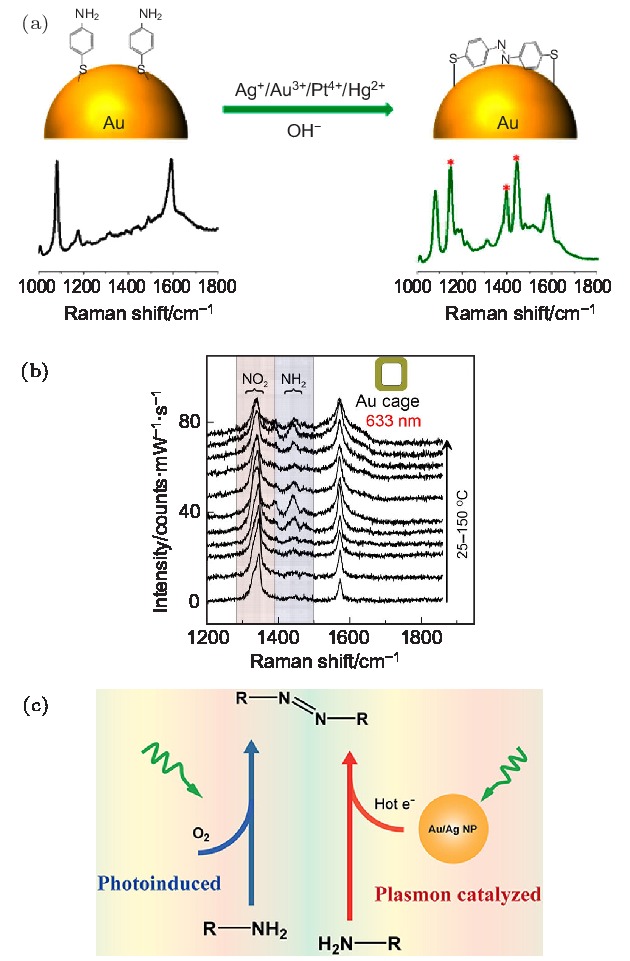
图 3 (a) Ag+, Au3+, Pt4+和Hg2+离子促进PATP分子在金纳米颗粒上发生聚合反应示意图及其产物的SERS光谱[34]; (b) 在升温条件下, 金纳米笼上PNTP分子的SERS光谱[15]; (c) D3ATP分子在光催化和等离激元热电子催化两种条件下的反应示意图[35]
Fig. 3. (a) Schematic diagram of Ag+, Au3+, Pt4+ and Hg2+ ions promoting the polymerization of PATP molecules on gold nanoparticle and SERS spectra of its products[34]; (b) SERS spectra of PNTP molecules on gold nanocages under elevated temperature conditions[15]; (c) schematic diagram of the reaction of D3ATP molecules under both photoinduced and plasmon hot electrons catalysis[35].
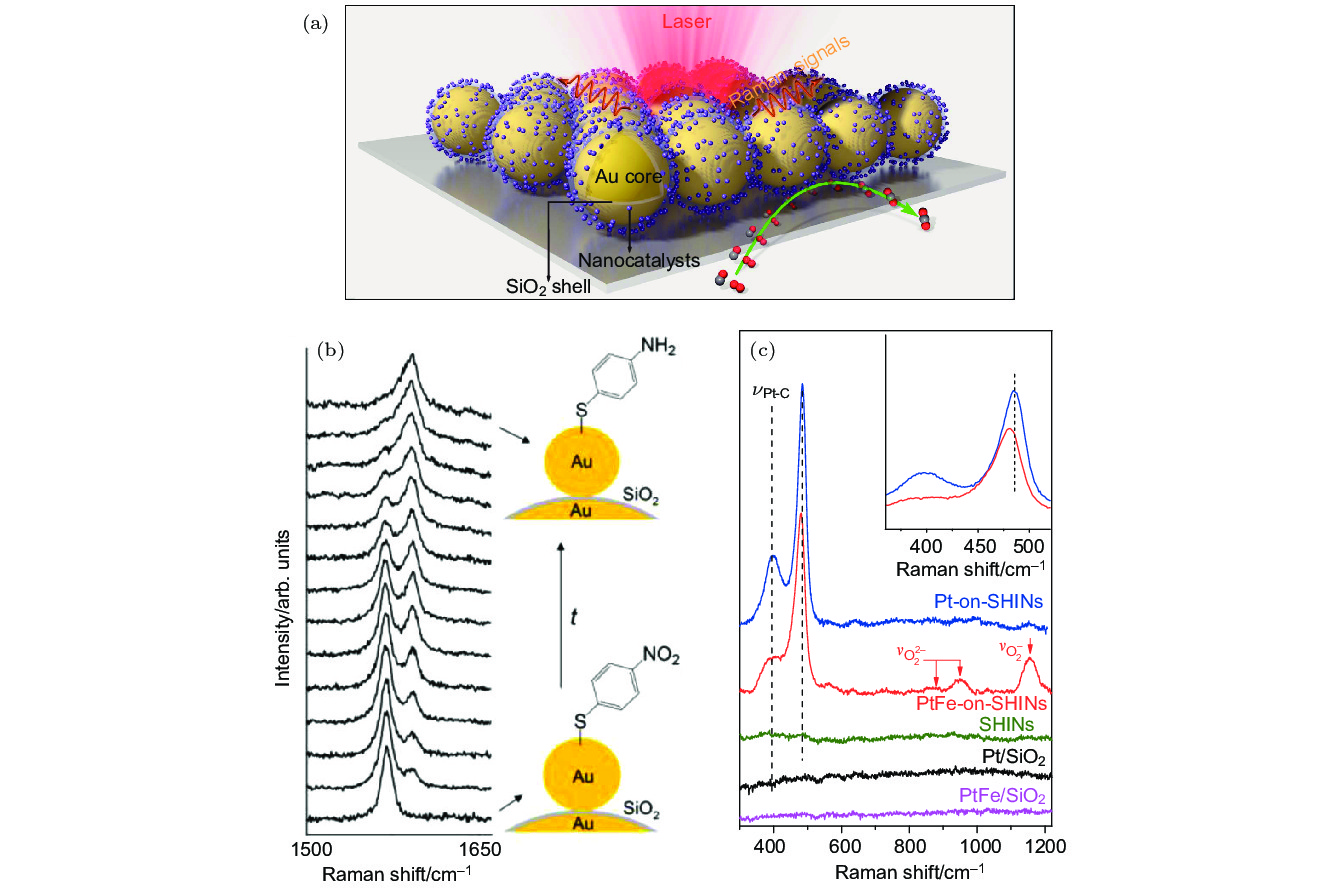
图 4 (a) Au NPs作为核心的壳层隔绝纳米颗粒卫星结构示意图[39]; (b) SHINERS技术监测的PNTP到PATP催化反应过程的原位拉曼光谱[38]; (c) SHINERS技术监测的PtFe-on-SHIN和Pt-on-SHIN纳米结构上CO氧化反应的原位拉曼光谱[39]
Fig. 4. (a) Schematic diagram of shell-isolated nanoparticle satellite structure with Au NPs core[39]; (b) in situ Raman spectroscopy of the PNTP to PATP catalytic reaction process monitored by SHINES technology[38]; (c) in situ Raman spectroscopy of CO oxidation reactions on PtFe-on-SHIN and Pt-on-SHIN nanostructures monitored by SHINERS technology[39].

图 5 (a) 单分子水平的PNTP到DMAB分子化学反应的测量示意图[47]; (b) 两个金纳米颗粒间单分子水平的PNTP到DMAB分子的催化示意图49]; (c) 在单分子水平等离激元催化PNTP分子的分解反应示意图; (d) 等离激元催化PNTP分子的时间依赖拉曼光谱[46]
Fig. 5. (a) The measurement schematic diagram of the molecular reaction of PNTP to DMAB at the single molecule level[47]; (b) schematic diagram of PNTP to DMAB molecular catalysis at single molecule level between two gold nanoparticles[49]; (c) schematic diagram of PNTP molecule decomposition at the single molecule level; (d) time-dependent Raman spectroscopy of PNTP molecular with plasmon catalysis[46].
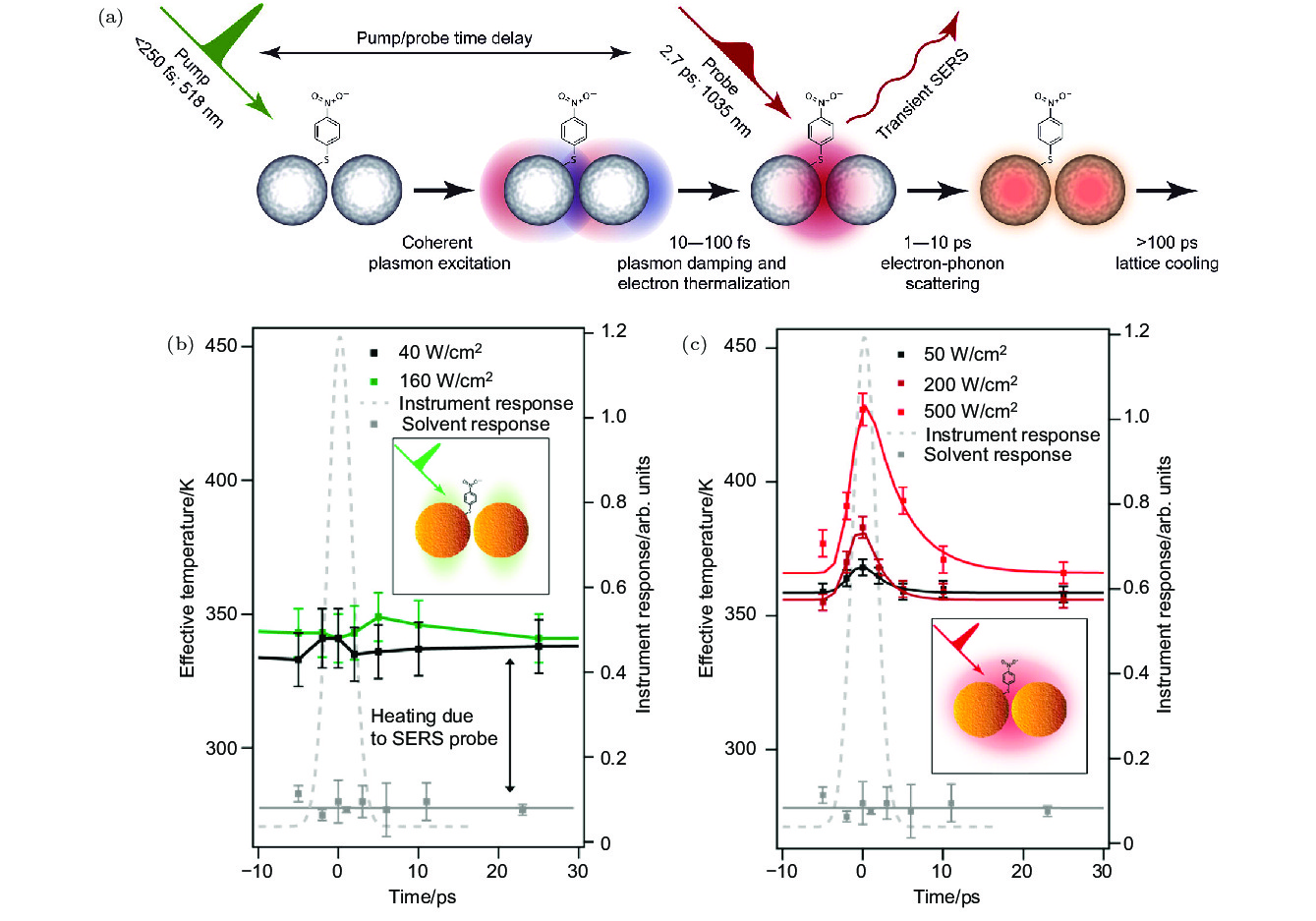
图 6 (a) 超快等离激元激发和驱动的分子动力学测量示意图[50]; (b) 在518 nm波长激光的激发下, 吸附到金纳米颗粒聚集体上的PNTP分子的有效温度与时间的依赖关系; (c) 在1035 nm波长激光的激发下, 吸附到金纳米颗粒聚集体上的PNTP分子的有效温度与时间的依赖关系[51]
Fig. 6. (a) Schematic diagram of molecular dynamics measurements of ultrafast plasmon excitation and driving[50]; (b) dependence of effective temperature on time of PNTP molecules adsorbed on gold nanoparticle aggregates under excitation of 518 nm laser; (c) dependence of effective temperature on time of PNTP molecules adsorbed on gold nanoparticle aggregates under excitation of 1035 nm laser[51].

图 7 (a) 大气TERS实验装置的示意图[52]; (b) 超高真空TERS实验装置的示意图[53]; (c) 使用TERS进行PtBL/Au(111)衬底上苯基异氰化物分子(PIC)测量的装置图[54]; (d) 4-氯苯基异氰化物分子(CPI)吸附在Pt(111), PtBL/Au(111)和PtML/Au(111)衬底上的TERS光谱[55]
Fig. 7. (a) Schematic diagram of an atmospheric TERS experimental device[52]; (b) schematic diagram of the ultra-high vacuum TERS experimental device[53]; (c) device diagram for the measurement of phenyl isocyanide molecules (PIC) on PtBL/Au(111) substrates[54]; (d) TERS spectra of 4-chlorophenyl isocyanide molecules (CPI) adsorbed on Pt(111), PtBL/Au(111) and PtML/Au(111) substrates[55].
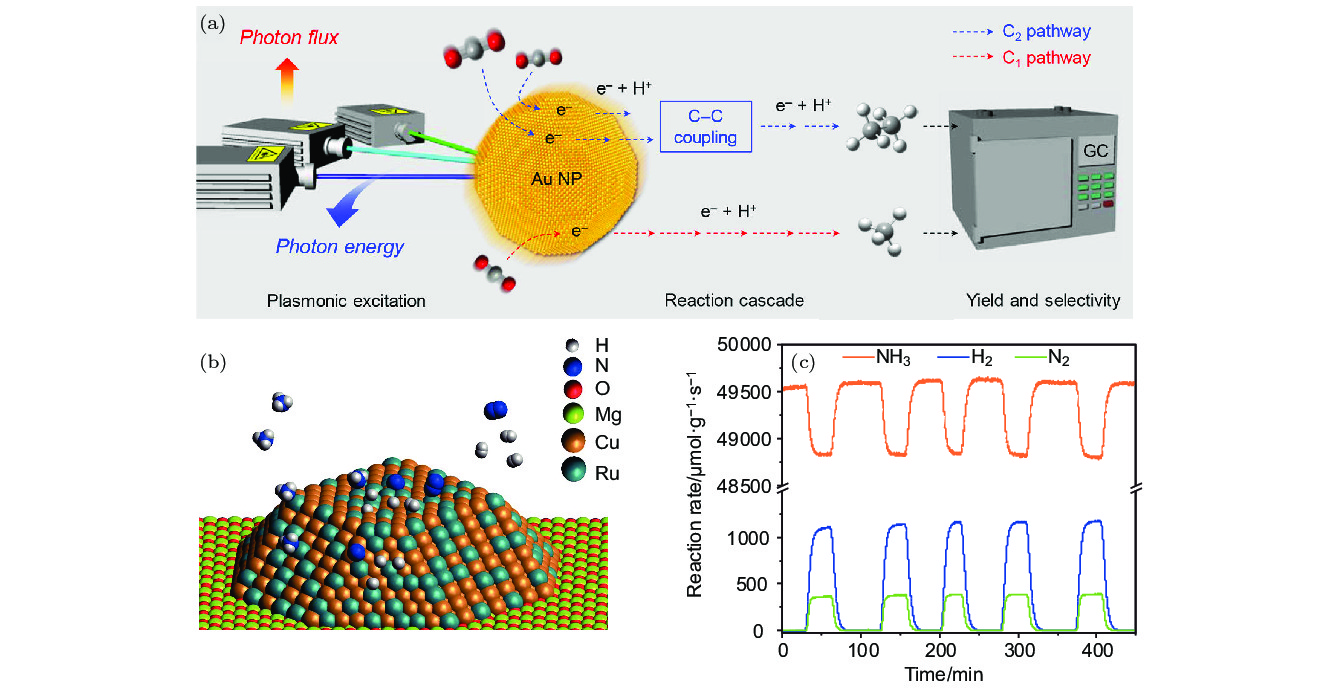
图 8 (a) Au NPs等离激元诱导的光催化CO2转化成烃的反应系统示意图, 其中通过GC监测反应产物与反应速率[59]; (b) 铜钌合金天线反应器(Cu-Ru-AR)的结构示意图, 其由Cu-Ru表面合金组成; (c) 利用GC监测获得的Cu-Ru-AR的光催化速率 (9.6 W/cm–2)[5]
Fig. 8. (a) Schematic diagram of plasmon-induced photocatalytic conversion of CO2 to hydrocarbon on the surface of Au NPs, wherein the reaction products and reaction rate are monitored by GC[59]; (b) schematic diagram of the structure of Cu-Ru-AR, which consists of a Cu-Ru surface alloy; (c) photocatalytic rate (9.6 W/cm–2) of Cu-Ru-AR monitored by GC[5].

图 9 (a) 等离激元催化仲-苯乙醇和苯甲醇的反应化学式以及在532 nm光照射下, 仲-苯乙醇反应前后的颜色变化; (b) 利用高效液相色谱紫外法(HPLC-UV)监测的光催化产物转化率, 激光照射次数依赖的苯乙酮转化百分比; (c) 利用HPLC-UV监测的光催化产物转化率, LED光照射时间依赖的苯乙酮和苯甲醛的转化百分比[60]
Fig. 9. (a) The chemical formula of the plasmon catalytic reaction of sec-phenethyl and benzyl alcohols, color change before and after the reaction of the sec-phenethyl alcohol under 532 nm light irradiation; (b) photocatalytic product conversion monitored by high performance liquid chromatography (HPLC-UV), the conversion percentage of thiophenone dependent on the number of laser irradiations; (c) photocatalytic product conversion monitored by HPLC-UV, conversion percentage of acetophenone and benzaldehyde dependent on LED light irradiation time[60].

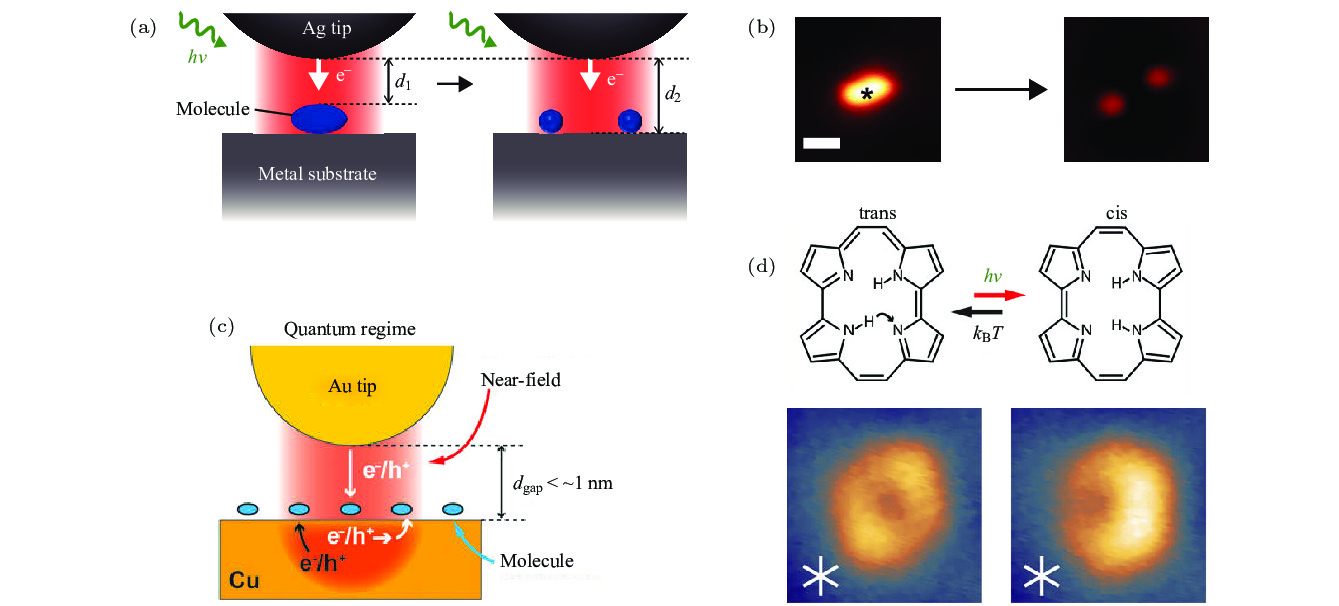
图 11 (a) STM监测等离激元诱导CH3S2分子分解示意图; (b) CH3S2分子分解前后的STM表征图[6]; (c) STM监测等离激元催化卟啉分子转变构型示意图; (d) 反式和顺式构型卟啉分子的化学结构和STM图像[62]
Fig. 11. (a) Schematic diagram of STM monitoring plasmon-induced CH3S2 molecular decomposition; (b) STM topography before and after decomposition of CH3S2 molecules[6]; (c) schematic diagram of STM monitoring plasmon catalyzed porphyrin molecular transformation configuration; (d) chemical structure and STM image of porphyrin molecules in trans and cis configuration[62].
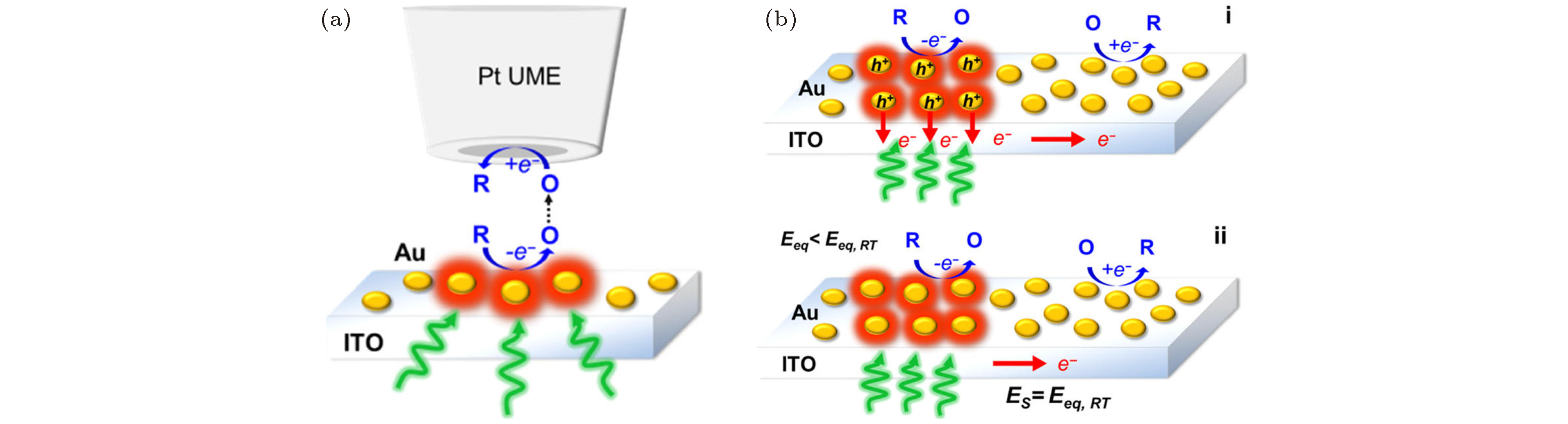
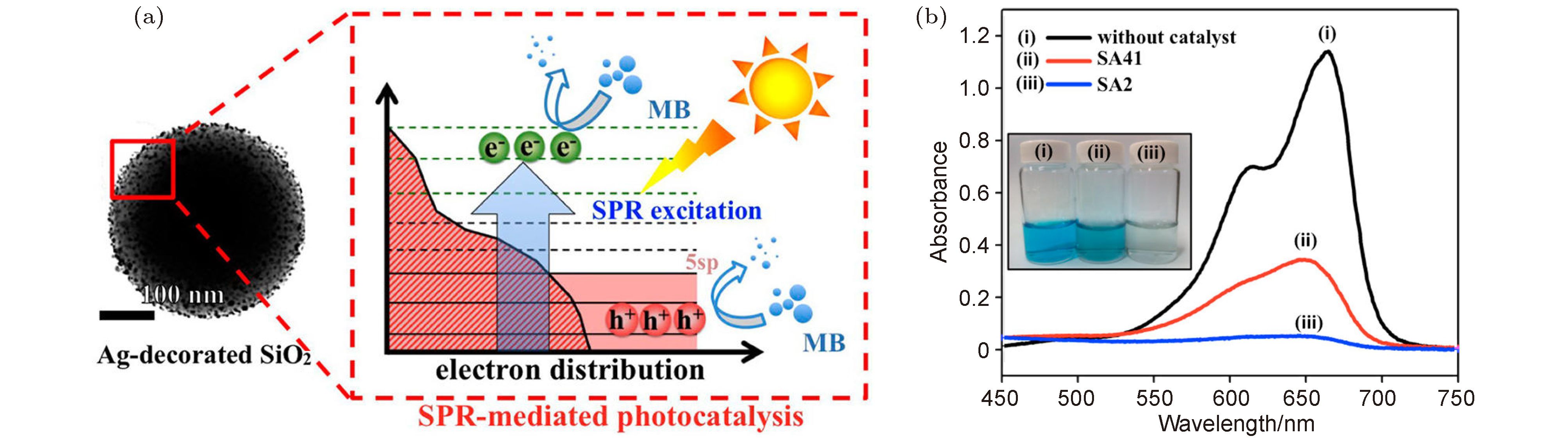
图 13 (a) 太阳光驱动的Ag NPs等离激元催化示意图; (b) 在太阳光照射下, 反应产物的紫外可见吸收光谱, 插图为对应溶液的颜色[64]
Fig. 13. (a) Schematic diagram of plasmon catalysis of Ag NPs driven by sunlight; (b) the UV-visible absorption spectrum of the reaction product under sunlight, and the inside illustration is the corresponding solution color[64].
-
[1] Zhang Z, Deckert G T, Deckert V 2015 Analyst 140 4325
Google Scholar
[2] Jiang R, Zhang M, Qian S L, Yan F, Pei L Q, Jin S, Zhao L B, Wu D Y, Tian Z Q 2016 J. Phys. Chem. C 120 16427
Google Scholar
[3] Tesema T E, Kafle B, Tadesse M G, Habteyes T G 2017 J. Phys. Chem. C 121 7421
[4] Zhang X, Li X, Zhang D, Su N Q, Yang W, Everitt H O, Liu J 2017 Nat. Commun. 8 14542
Google Scholar
[5] Zhou L, Swearer D F, Zhang C, Robatjazi H, Zhao H, Henderson L, Dong L, Christopher P, Carter E A, Nordlander P, Halas N J 2018 Science 362 69
Google Scholar
[6] Kazuma E, Jung J, Ueba H, Trenary M, Kim Y 2018 Science 360 521
Google Scholar
[7] Zhan C, Chen X J, Yi J, Li J F, Wu D Y, Tian Z Q 2018 Nat. Rev. Chem. 2 216
Google Scholar
[8] Liu Z, Hou W, Pavaskar P, Aykol M, Cronin S B 2011 Nano Lett. 11 1111
Google Scholar
[9] Chen J, Bailey C S, Hong Y, Wang L, Cai Z, Shen L, Hou B, Wang Y, Shi H, Sambur J, Ren W, Pop E, Cronin S B 2019 ACS Photonics 6 787
Google Scholar
[10] Kazuma E, Kim Y 2019 Angew. Chem. Int. Ed. 58 4800
Google Scholar
[11] Xie W, Schlücker S 2015 Nat. Commun. 6 7570
Google Scholar
[12] Zhang Y, Nelson T, Tretiak S, Guo H, Schatz G C 2018 ACS Nano 12 8415
Google Scholar
[13] Wu K, Chen J, McBride J R, Lian T 2015 Science 349 632
Google Scholar
[14] Duchene J S, Tagliabue G, Welch A J, Cheng W H, Atwater H A 2018 Nano Lett. 18 2545
Google Scholar
[15] Golubev A A, Khlebtsov B N, Rodriguez R D, Chen Y, Zahn D R T 2018 J. Phys. Chem. C 122 5657
[16] Zhang X, Li X, Reish M E, Zhang D, Su N Q, Gutiérrez Y, Moreno F, Yang W, Everitt H O, Liu J 2018 Nano Lett. 18 1714
Google Scholar
[17] Jeanmaire D L, Van R P 1977 J. Electroanal. Chem. Interfacial Electrochem. 84 1
Google Scholar
[18] Albrecht M G, Creighton J A 1977 J. Am. Chem. Soc. 99 5215
Google Scholar
[19] Kleinman S L, Frontiera R R, Henry A I, Dieringer J A, Duyne R P V 2012 Phys. Chem. Chem. Phys. 15 21
[20] Zhang C, Jiang S Z, Huo Y Y, Liu A H, Xu S C, Liu X Y, Sun Z C, Xu Y Y, Li Z, Man B Y 2015 Opt. Express 23 24811
Google Scholar
[21] Cao E, Lin W, Sun M, Liang W, Song Y 2018 Nanophotonics 7 145
Google Scholar
[22] Li C, Yu J, Xu S, Jiang S, Xiu X, Chen C, Liu A, Wu T, Man B, Zhang C 2018 Adv. Mater. Technol. 3 1800174
Google Scholar
[23] Xu J, Li C, Si H, Zhao X, Wang L, Jiang S, Wei D, Yu J, Xiu X, Zhang C 2018 Opt. Express 26 21546
Google Scholar
[24] Zhang C, Li C, Yu J, Jiang S, Xu S, Yang C, Liu Y J, Gao X, Liu A, Man B 2018 Sens. Actuators B: Chem. 258 163
Google Scholar
[25] Zhang Z, Sheng S, Wang R, Sun M 2016 Anal. Chem. 88 9328
Google Scholar
[26] Anderson M S 2000 Appl. Phys. Lett. 76 3130
Google Scholar
[27] Hayazawa N, Inouye Y, Sekkat Z, Kawata S 2000 Opt. Commun. 183 333
Google Scholar
[28] Stöckle R M, Suh Y D, Deckert V, Zenobi R 2000 Chem. Phys. Lett. 318 131
Google Scholar
[29] Verma P 2017 Chem. Rev. 117 6447
Google Scholar
[30] Han S W, Lee I, Kim K 2002 Langmuir 18 182
Google Scholar
[31] Huang Y F, Zhu H P, Liu G K, Wu D Y, Ren B, Tian Z Q 2010 J. Am. Chem. Soc. 132 9244
Google Scholar
[32] Fang Y, Li Y, Xu H, Sun M 2010 Langmuir 26 7737
Google Scholar
[33] Dong B, Fang Y, Chen X, Xu H, Sun M 2011 Langmuir 27 10677
Google Scholar
[34] Zhang Z, Merk V, Hermanns A, Unger W E S, Kneipp J 2017 ACS Catal. 7 7803
Google Scholar
[35] Zhang Z, Kinzel D, Deckert V 2016 J. Phys. Chem. C 120 20978
Google Scholar
[36] Li J F, Huang Y F, Ding Y, Yang Z L, Li S B, Zhou X S, Fan F R, Zhang W, Zhou Z Y, Wu D Y, Ren B, Wang Z L, Tian Z Q 2010 Nature 464 392
Google Scholar
[37] Wang Y H, Wei J, Radjenovic P, Tian Z Q, Li J F 2019 Anal. Chem. 91 1675
Google Scholar
[38] Xie W, Walkenfort B, Schlücker S 2013 J. Am. Chem. Soc. 135 1657
Google Scholar
[39] Zhang H, Wang C, Sun H L, Fu G, Chen S, Zhang Y J, Chen B H, Anema J R, Yang Z L, Li J F, Tian Z Q 2017 Nat. Commun. 8 15447
Google Scholar
[40] Kneipp K, Wang Y, Kneipp H, Perelman L T, Itzkan I, Dasari R R, Feld M S 1997 Phys. Rev. Lett. 78 1667
Google Scholar
[41] Nie S, Emory S R 1997 Science 275 1102
Google Scholar
[42] Chen H Y, Lin M H, Wang C Y, Chang Y M, Gwo S 2015 J. Am. Chem. Soc. 137 13698
Google Scholar
[43] Marshall A R L, Stokes J, Viscomi F N, Proctor J E, Gierschner J, Bouillard J S G, Adawi A M 2017 Nanoscale 9 17415
Google Scholar
[44] Kim N H, Hwang W, Baek K, Rohman M R, Kim J, Kim H W, Mun J, Lee S Y, Yun G, Murray J, Ha J W, Rho J, Moskovits M, Kim K 2018 J. Am. Chem. Soc. 140 4705
Google Scholar
[45] Santos D P, Temperini M L A, Brolo A G 2019 Acc. Chem. Res. 52 456
Google Scholar
[46] Zhang Z, Deckert G T, Singh P, Deckert V 2015 Chem. Commun. 51 3069
Google Scholar
[47] Choi H K, Park W H, Park C G, Shin H H, Lee K S, Kim Z H 2016 J. Am. Chem. Soc. 138 4673
Google Scholar
[48] Sprague E A, McAnally M O, Zhdanov D V, Zrimsek A B, Apkarian V A, Seideman T, Schatz G C, van Duyne R P 2017 J. Am. Chem. Soc. 139 15212
Google Scholar
[49] Brooks J L, Frontiera R R 2016 J. Phys. Chem. C 120 20869
Google Scholar
[50] Brandt N C, Keller E L, Frontiera R R 2016 J. Phys. Chem. Lett. 7 3179
Google Scholar
[51] Keller E L, Frontiera R R 2018 ACS Nano 12 5848
Google Scholar
[52] Schrojenstein E M, Deckert G T, Mank A J G, Deckert V, Weckhuysen B M 2012 Nat. Nanotechnol. 7 583
Google Scholar
[53] Sun M, Zhang Z, Zheng H, Xu H 2012 Sci. Rep. 2 647
Google Scholar
[54] Zhong J H, Jin X, Meng L, Wang X, Su H S, Yang Z L, Williams C T, Ren B 2017 Nat. Nanotechnol. 12 132
Google Scholar
[55] Su H S, Zhang X G, Sun J J, Jin X, Wu D Y, Lian X B, Zhong J H, Ren B 2018 Angew. Chem. Int. Ed. 57 13177
Google Scholar
[56] Zhang Z, Sheng S, Zheng H, Xu H, Sun M 2014 Nanoscale 6 4903
Google Scholar
[57] Zhang Z, Chen L, Sun M, Ruan P, Zheng H, Xu H 2013 Nanoscale 5 3249
Google Scholar
[58] Zhang Z, Richard L M, Deckert V 2017 Faraday Discuss. 205 213
Google Scholar
[59] Yu S, Wilson A J, Heo J, Jain P K 2018 Nano Lett. 18 2189
Google Scholar
[60] Hallett G L, Silvero M J, González M, Grenier M, Netto J C, Scaiano J C 2011 J. Phys. Chem. C 115 10784
Google Scholar
[61] Vadai M, Angell D K, Hayee F, Sytwu K, Dionne J A 2018 Nat. Commun. 9 4658
Google Scholar
[62] Böckmann H, Gawinkowski S, Waluk J, Raschke M B, Wolf M, Kumagai T 2018 Nano Lett. 18 152
Google Scholar
[63] Yu Y, Sundaresan V, Willets K A 2018 J. Phys. Chem. C 122 5040
Google Scholar
[64] Chen K H, Pu Y C, Chang K D, Liang Y F, Liu C M, Yeh J W, Shih H C, Hsu Y J 2012 J. Phys. Chem. C 116 19039
Google Scholar

 下载:
下载:
计量
- 文章访问数: 27927
- PDF下载量: 500
- 被引次数: 0
