










-
通过详尽的第一性原理计算, 提出了一类新型的二维III族金属硫族化合物MX (M = Al, Ga, In; X = S, Se, Te)的同素异形体. 这类化合物的结构是由正方形和八边形环构成的. 计算得到的结合能和声子谱表明, 所有的结构都同时具有能量和动力学稳定性. 所有结构都是间接带隙半导体, 其带隙大小随X原子由S到Se到Te的变化而减小. 计算结果表明这类材料具有很广的带隙范围, 从1.88到3.24 eV, 同时它们的能带结构可以通过双轴应变进一步调节. 这些结构具有丰富的电子结构性质和可调的带隙, 有可能被用于未来纳米电子学领域.
-
关键词:
- III族金属硫族化合物 /
- 电子结构 /
- 第一性原理计算
Single-layered III-VI compounds have potential applications in many fields, such as highly sensitive photodetectors, field effect transistors, and electrochemical sensors, due to their wide range photosensitivities and excellent electronic properties. This paper presents a new two-dimensional tetragonal allotrope (called haeckelites structure) of single layered group III monochalcogenides MX (M = Al, Ga, In; X = S, Se, Te), which are constructed from the square and octagon rings. The first-principles calculations are performed using the Vienna ab initio simulation package (VASP) based on density functional theory (DFT). The cohesive energy of the haeckelite structure MX is positive and a little smaller than that (0.07—0.10 eV) of the hexagonal MX. The phonon spectra for the haeckelites structure MX have basically no imaginary frequencies in the whole Brillouin zone. The calculated binding energy and phonon spectrum show that these structures are energetically and dynamically stable. For all the compounds, the charge density isosurfaces show that most electrons are localized at the positions of X and M atoms, indicating that the M—X bond is ionic and M—M bond is covalent. All of haeckelite structure MX are indirect bandgap semiconductors, and their band gap sizes decrease with the X atom changing from S to Se to Te. For example, the band gaps of InS, InSe, and InTe are 2.42, 2.07, and 1.88 eV, respectively. The calculation results show that these materials have a wide band gap range from 1.88 to 3.24 eV. We find that the band gaps of AlS, AlSe, and GaS are relatively large with the values of 3.08, 3.03, and 3.24 eV, respectively. This may make them suitable for optically transparent devices. The band structures of GaSe, InS, InSe, and InTe can be further modulated by the biaxial strains. Their band gaps decrease linearly with the strain increasing. The band gap of AlS and AlSe both first increase and then decrease with the strain increasing.[1] Late D J, Liu B, Luo J, Yan A M, Matte H S S R, Grayson M, Rao C N R, Dravid V P 2012 Adv. Mater. 24 3549
 Google Scholar
Google Scholar
[2] Hu P, Wang L, Yoon M, Zhang J, Feng W, Wang X, Wen Z, Idrobo J C, Miyamoto Y, Geohegan D B, Xiao K 2013 Nano Lett. 13 1649
Google Scholar
[3] Kuhn A, Chevy A, Chevalier R 1975 Phys. Status Solidi A 31 469
Google Scholar
[4] Hu P, Wen Z, Wang L, Tan P, Xiao K 2012 ACS Nano 6 5988
Google Scholar
[5] Lei S, Ge L, Liu Z, Najmaei S, Shi G, You G, Lou J, Vajtai R, Ajayan P M 2013 Nano Lett. 13 2777
Google Scholar
[6] Zhou Y, Nie Y, Liu Y, Yan K, Hong J, Jin C, Zhou Y, Yin J, Liu Z, Peng H 2014 ACS Nano 8 1485
Google Scholar
[7] Mudd G W, Svatek S A, Hague L, et al. 2015 Adv. Mater. 27 3760
Google Scholar
[8] Yang S, Li Y, Wang X, Huo N, Xia J B, Li S S, Li J 2014 Nanoscale 6 2582
Google Scholar
[9] Zhuang H L, Hennig R G 2013 Chem. Mater. 25 3232
Google Scholar
[10] Cao T, Li Z, Louie S G 2015 Phys. Rev. Lett. 114 236602
Google Scholar
[11] Li W, Li J 2015 Nano Res. 8 3796
Google Scholar
[12] Wypych F, Schöllhorn R 1992 J. Chem. Soc., Chem. Commun. 138 6
[13] Tang Q, Jiang D E 2015 Chem. Mater. 27 3743
Google Scholar
[14] Terrones H, Terrones M, Hernández E, Grobert N, Charlier J C, Ajayan P M 2000 Phys. Rev. Lett. 84 1716
Google Scholar
[15] Enyashin A N, Ivanovskii A L 2011 Phys. Status Solidi B 248 1879
Google Scholar
[16] Liu Y, Wang G, Huang Q, Guo L, Chen X 2012 Phys. Rev. Lett. 108 225505
Google Scholar
[17] Sun H, Mukherjee S, Daly M, Krishnan A, Karigerasi M H, Singh C V 2016 Carbon 110 443
Google Scholar
[18] Li W, Guo M, Zhang G, Zhang Y W 2014 Phys. Rev. B 89 205402
Google Scholar
[19] Ma Y, Kou L, Li X, Dai Y, Smith S C, Heine T 2015 Phys. Rev. B 92 085427
Google Scholar
[20] Nie S M, Song Z, Weng H, Fang Z 2015 Phys. Rev. B 91 235434
Google Scholar
[21] Sun Y, Felser C, Yan B 2015 Phys. Rev. B 92 165421
Google Scholar
[22] Ersan F, Aktürk E, Ciraci S 2016 Phys. Rev. B 94 245417
Google Scholar
[23] Kou L, Tan X, Ma Y, Tahini H, Zhou L, Sun Z, Aijun D, Chen C, Smith S C 2015 2D Mater. 2 045010
[24] Kresse G, Furthmüller J 1996 Phys. Rev. B 54 11169
Google Scholar
[25] Kresse G, Furthmüller J 1996 Comp. Mater. Sci. 6 15
Google Scholar
[26] Perdew J P, Burke K, Ernzerhof M 1996 Phys. Rev. Lett. 77 3865
Google Scholar
[27] Blöchl P E 1994 Phys. Rev. B 50 17953
Google Scholar
[28] Kresse G, Joubert D 1999 Phys. Rev. B 59 1758
[29] Monkhorst H J, Pack J D 1976 Phys. Rev. B 13 5188
Google Scholar
[30] Paier J, Marsman M, Hummer K, Kresse G, Gerber I C, Ángyán J G 2006 J. Chem. Phys. 124 154709
Google Scholar
[31] Togo A, Oba F, Tanaka I 2008 Phys. Rev. B 78 134106
Google Scholar
[32] Demirci S, Avazlı N, Durgun E, Cahangirov S 2017 Phys. Rev. B 95 115409
Google Scholar
[33] Zólyomi V, Drummond N D, Fal'ko V I 2014 Phys. Rev. B 89 205416
Google Scholar
[34] Zheng H, Li X B, Chen N K, Xie S Y, Tian W Q, Chen Y, Xia H, Zhang S B, Sun H B 2015 Phys. Rev. B 92 115307
Google Scholar
[35] Monroy E, Omn s F, Calle F 2003 Semicond. Sci. Technol. 18 R33
Google Scholar
[36] Caraveo Frescas J A, Nayak P K, Al Jawhari H A, Granato D B, Schwingenschlögl U, Alshareef H N 2013 ACS Nano 7 5160
Google Scholar
-
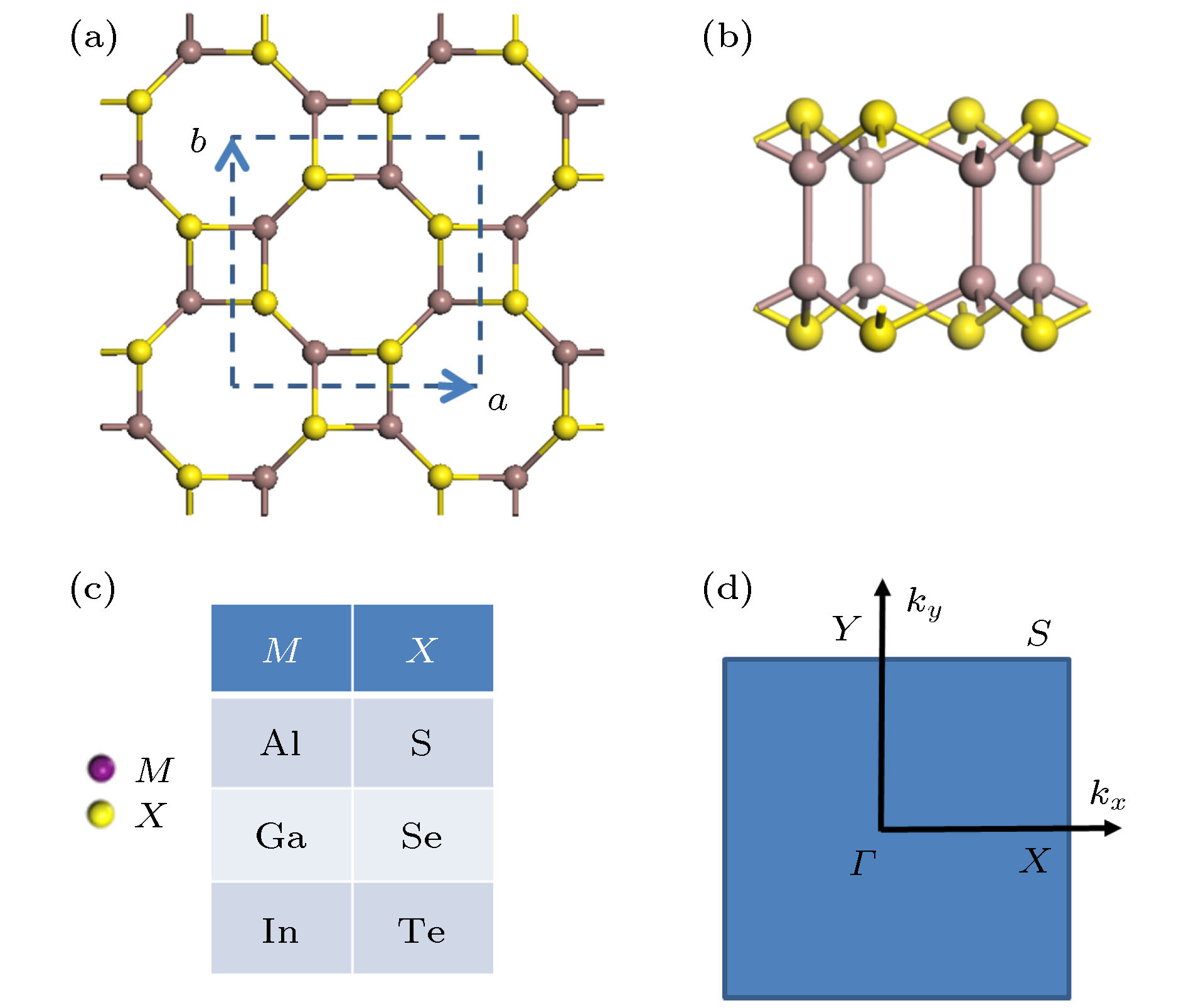
图 1 四方III族金属硫族化合物MX的几何结构 (a), (b) 分别对应原子结构的俯视图和侧视图; (c) 体系计算中选择的各种组成成分; (d) 体系的第一布里渊区, 其中字母标出了能带计算时的特殊点和路径
Fig. 1. The geometric structures of tetragonal group III monochalcogenide MX: (a) and (b) Top-view and side-view of the structure; (c) considered systems with various choice of compositions; (d) the first Brillouin zone of structure with letters designating special points and the lines for band-structure calculation.
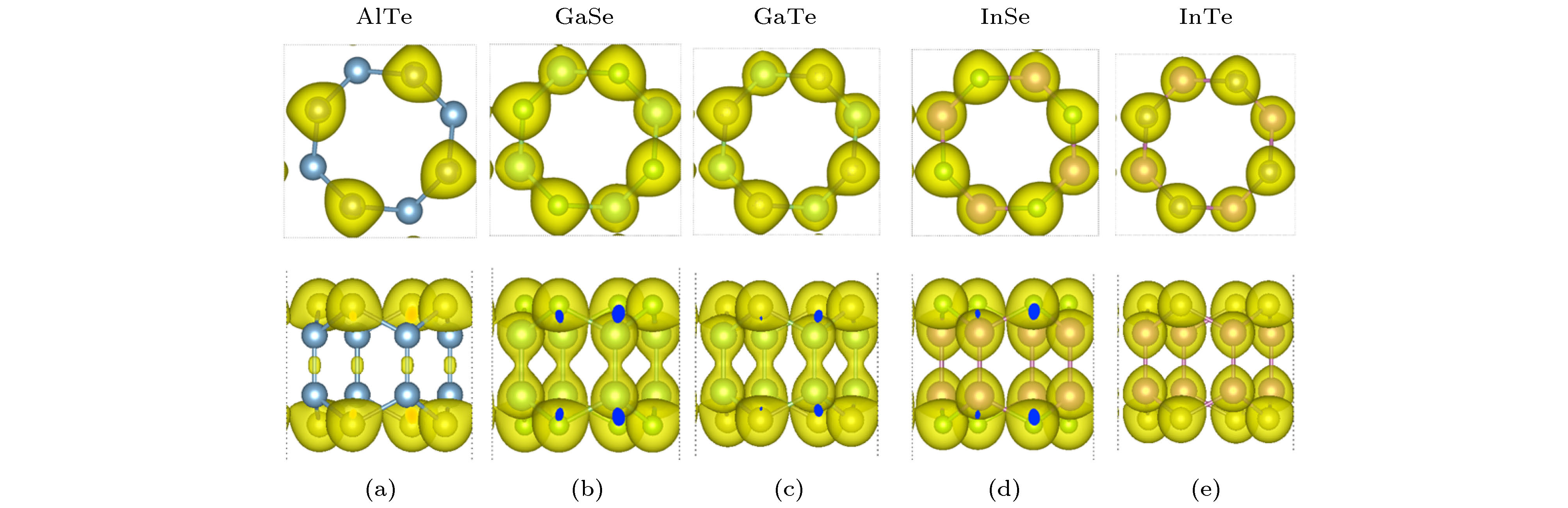
图 2 四方MX的总电荷密度等值面的俯视图和侧视图 (a) AlTe; (b) GaSe; (c) GaTe; (d) InSe; (e) InTe. 选择的总电荷密度等值面的值为0.05 electrons/Å3
Fig. 2. Top view and side view of the total charge density isosorfaces of tetragonal MX: (a) AlTe; (b) GaSe; (c) GaTe; (d) InSe; (e) InTe. Isosurface value is 0.05 electrons/Å3.

图 3 单层haeckelites结构III族金属硫族化合物沿着二维布里渊区高对称方向的振动声子谱 (a) AlS; (b) AlSe; (c) AlTe; (d) GaS; (e) GaSe; (f) GaTe; (g) InS; (h) InSe; (i) InTe
Fig. 3. Vibrational phonon spectra of group III monochalocogenide haeckelites monolayers along the high symmetry directions in the 2D Brillouin zone: (a) AlS; (b) AlSe; (c) AlTe; (d) GaS; (e) GaSe; (f) GaTe; (g) InS; (h) InSe; (i) InTe.
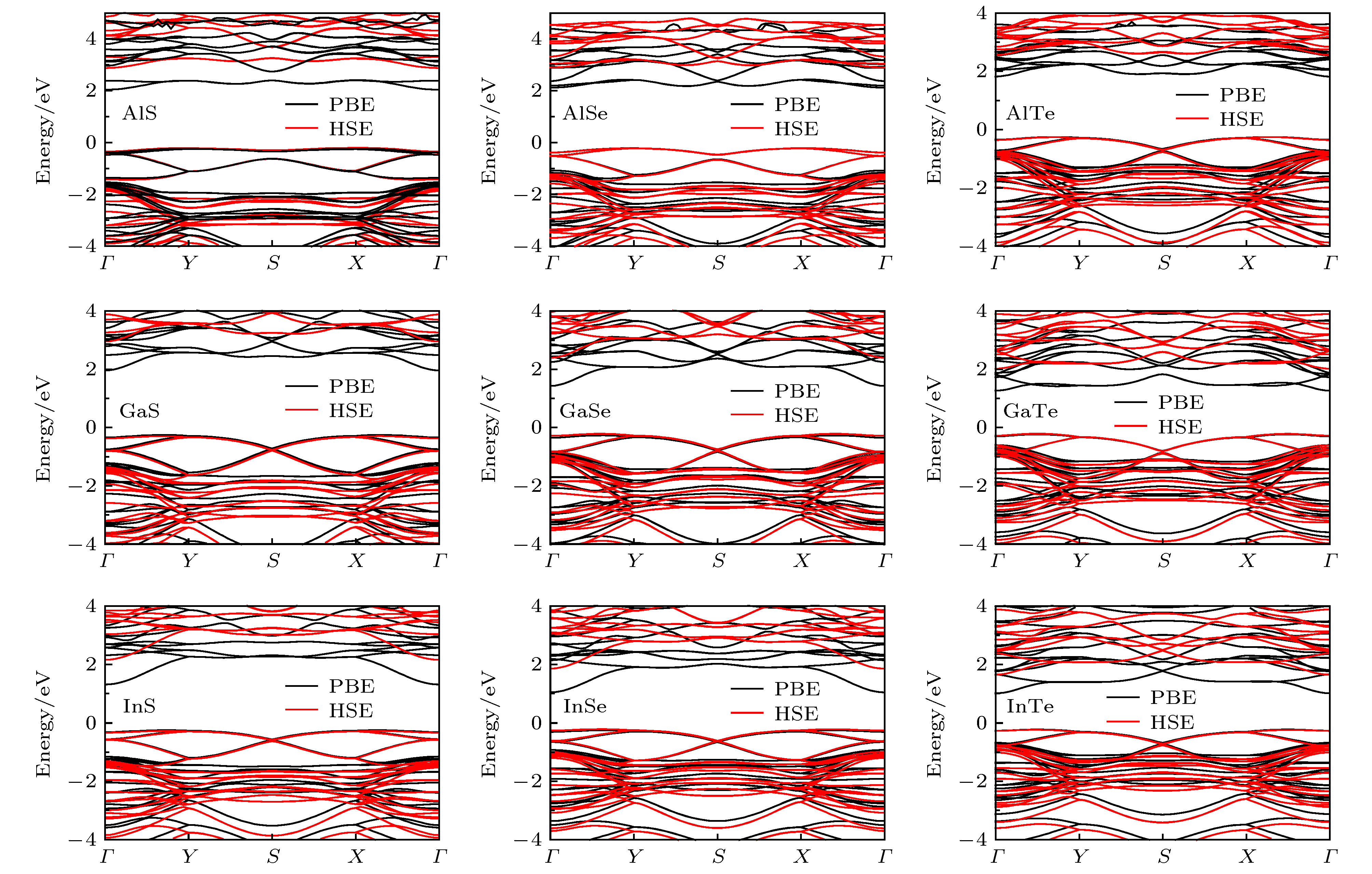
图 4 PBE和HSE泛函给出的四方MX单层的能带结构, 所有化合物都是间接带隙半导体
Fig. 4. The PBE and HSE band structures for tetragonal MX monolayers. All the compounds are indirect band gap semiconductors.
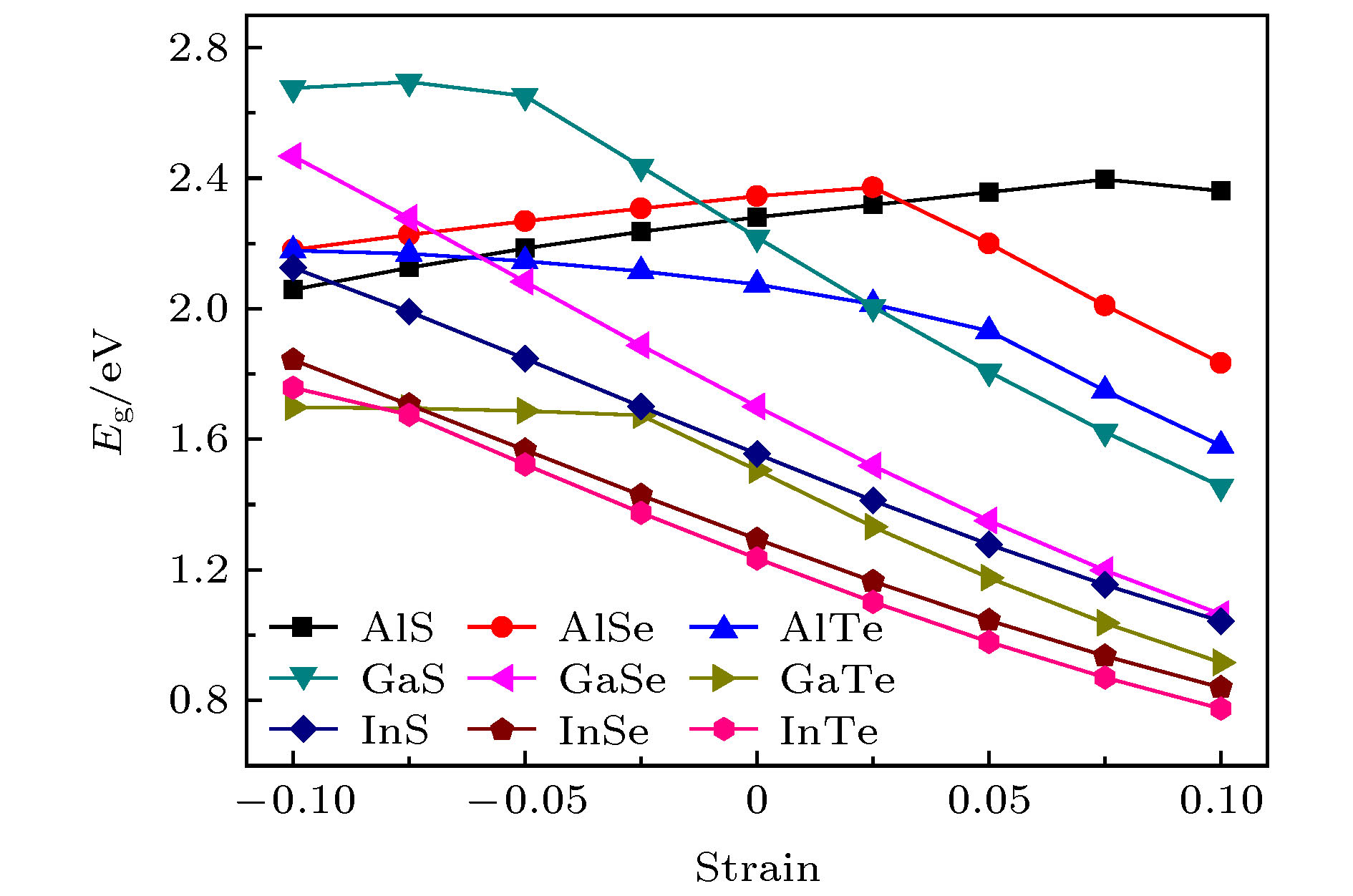
图 5 四方MX在不同的层内应变强度(–0.10—0.10)下的电子带隙
Fig. 5. The electronic band gaps for tetragonal MX as a function of the in-layer strain from –0.10 to 0.10.
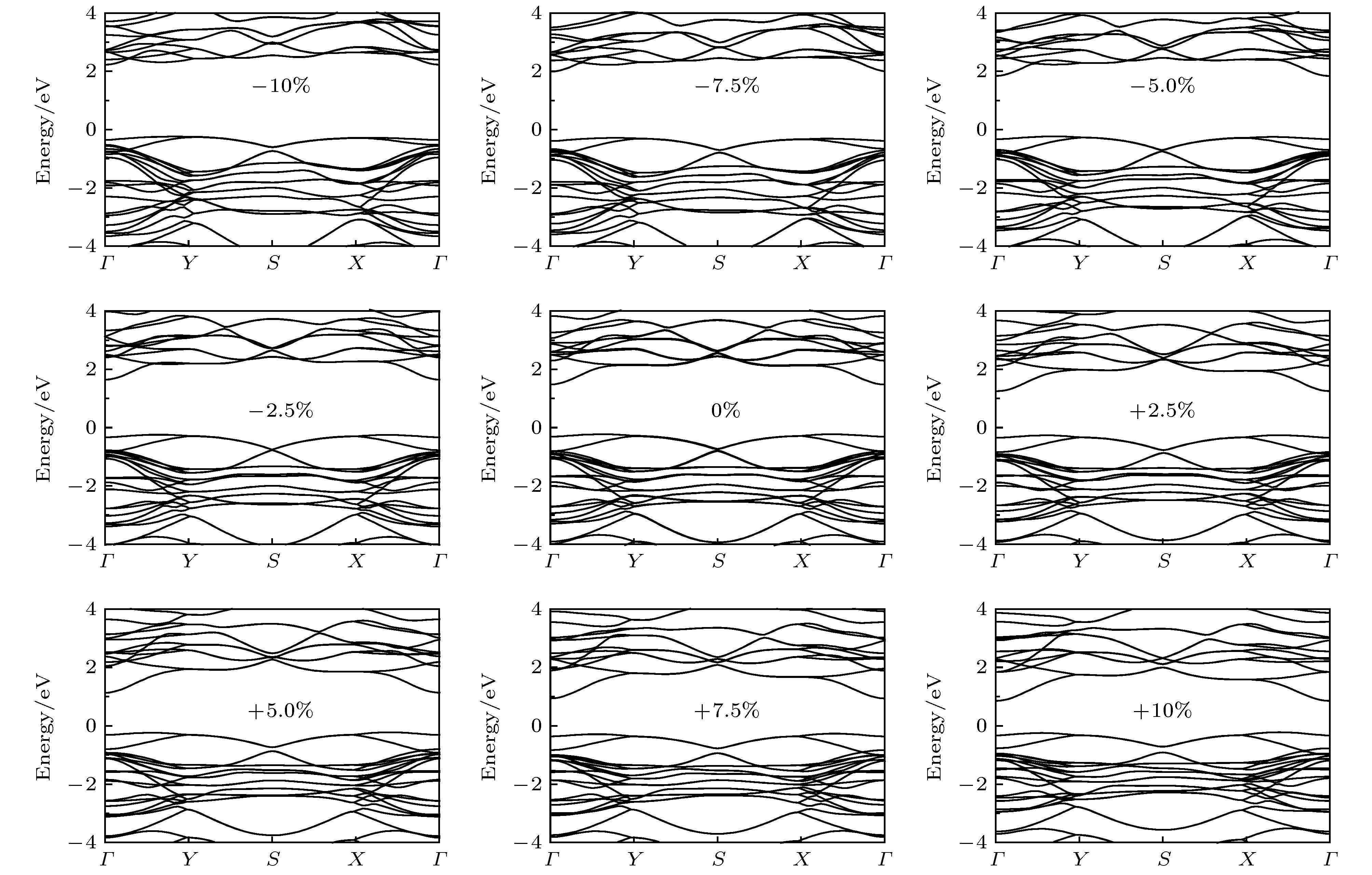
图 6 二维四方GaSe在不同应变下的电子能带结构, 其中费米能级设为零
Fig. 6. Electronic band structure of 2D tetragonal GaSe under different strains. The Fermi level is set to zero.
表 1 计算得到的二维四方III族金属硫族化合物MX (M = Al, Ga, In; X = S, Se, Te)的结构和电学性质: 晶格参数a和b, 结合能(EC)和分别用PBE和HSE06泛函计算得到的带隙(EgPBE和EgHSE), 表中同时给出了六方结构的结合能(EC(2H))和带隙(Eg(2H)PBE)
Table 1. The calculated structural and electronic properties of 2D tetragonal group III monochalcogenides MX (M = Al, Ga, and In, X = S, Se, and Te): Lattice parameters a and b, cohesive energy (EC), band gap calculated using PBE and HSE06 functional (EgPBE and EgHSE, respectively). For comparison, the cohesive energy (EC(2H)) and band gap (Eg(2H)PBE) of the hexagonal MX are also given.
MX a/Å b/Å EC/eV EC(2H)/eV EgPBE/eV EgHSE/eV Eg(2H)PBE/eV AlS 7.07 7.06 4.44 4.54 2.28 3.08 2.10 AlSe 7.43 7.45 3.98 4.07 2.34 3.03 1.99 AlTe 8.07 8.08 3.48 3.55 2.07 2.70 1.84 GaS 7.16 7.15 3.78 3.89 2.21 3.24 2.35 GaSe 7.51 7.53 3.42 3.52 1.70 2.61 1.77 GaTe 8.12 8.15 3.03 3.11 1.50 2.23 1.43 InS 7.76 7.75 3.49 3.59 1.55 2.42 1.64 InSe 8.08 8.09 3.18 3.27 1.29 2.07 1.37 InTe 8.64 8.67 2.83 2.91 1.23 1.88 1.29  下载: 导出CSV
下载: 导出CSV
-
[1] Late D J, Liu B, Luo J, Yan A M, Matte H S S R, Grayson M, Rao C N R, Dravid V P 2012 Adv. Mater. 24 3549
Google Scholar
[2] Hu P, Wang L, Yoon M, Zhang J, Feng W, Wang X, Wen Z, Idrobo J C, Miyamoto Y, Geohegan D B, Xiao K 2013 Nano Lett. 13 1649
Google Scholar
[3] Kuhn A, Chevy A, Chevalier R 1975 Phys. Status Solidi A 31 469
Google Scholar
[4] Hu P, Wen Z, Wang L, Tan P, Xiao K 2012 ACS Nano 6 5988
Google Scholar
[5] Lei S, Ge L, Liu Z, Najmaei S, Shi G, You G, Lou J, Vajtai R, Ajayan P M 2013 Nano Lett. 13 2777
Google Scholar
[6] Zhou Y, Nie Y, Liu Y, Yan K, Hong J, Jin C, Zhou Y, Yin J, Liu Z, Peng H 2014 ACS Nano 8 1485
Google Scholar
[7] Mudd G W, Svatek S A, Hague L, et al. 2015 Adv. Mater. 27 3760
Google Scholar
[8] Yang S, Li Y, Wang X, Huo N, Xia J B, Li S S, Li J 2014 Nanoscale 6 2582
Google Scholar
[9] Zhuang H L, Hennig R G 2013 Chem. Mater. 25 3232
Google Scholar
[10] Cao T, Li Z, Louie S G 2015 Phys. Rev. Lett. 114 236602
Google Scholar
[11] Li W, Li J 2015 Nano Res. 8 3796
Google Scholar
[12] Wypych F, Schöllhorn R 1992 J. Chem. Soc., Chem. Commun. 138 6
[13] Tang Q, Jiang D E 2015 Chem. Mater. 27 3743
Google Scholar
[14] Terrones H, Terrones M, Hernández E, Grobert N, Charlier J C, Ajayan P M 2000 Phys. Rev. Lett. 84 1716
Google Scholar
[15] Enyashin A N, Ivanovskii A L 2011 Phys. Status Solidi B 248 1879
Google Scholar
[16] Liu Y, Wang G, Huang Q, Guo L, Chen X 2012 Phys. Rev. Lett. 108 225505
Google Scholar
[17] Sun H, Mukherjee S, Daly M, Krishnan A, Karigerasi M H, Singh C V 2016 Carbon 110 443
Google Scholar
[18] Li W, Guo M, Zhang G, Zhang Y W 2014 Phys. Rev. B 89 205402
Google Scholar
[19] Ma Y, Kou L, Li X, Dai Y, Smith S C, Heine T 2015 Phys. Rev. B 92 085427
Google Scholar
[20] Nie S M, Song Z, Weng H, Fang Z 2015 Phys. Rev. B 91 235434
Google Scholar
[21] Sun Y, Felser C, Yan B 2015 Phys. Rev. B 92 165421
Google Scholar
[22] Ersan F, Aktürk E, Ciraci S 2016 Phys. Rev. B 94 245417
Google Scholar
[23] Kou L, Tan X, Ma Y, Tahini H, Zhou L, Sun Z, Aijun D, Chen C, Smith S C 2015 2D Mater. 2 045010
[24] Kresse G, Furthmüller J 1996 Phys. Rev. B 54 11169
Google Scholar
[25] Kresse G, Furthmüller J 1996 Comp. Mater. Sci. 6 15
Google Scholar
[26] Perdew J P, Burke K, Ernzerhof M 1996 Phys. Rev. Lett. 77 3865
Google Scholar
[27] Blöchl P E 1994 Phys. Rev. B 50 17953
Google Scholar
[28] Kresse G, Joubert D 1999 Phys. Rev. B 59 1758
[29] Monkhorst H J, Pack J D 1976 Phys. Rev. B 13 5188
Google Scholar
[30] Paier J, Marsman M, Hummer K, Kresse G, Gerber I C, Ángyán J G 2006 J. Chem. Phys. 124 154709
Google Scholar
[31] Togo A, Oba F, Tanaka I 2008 Phys. Rev. B 78 134106
Google Scholar
[32] Demirci S, Avazlı N, Durgun E, Cahangirov S 2017 Phys. Rev. B 95 115409
Google Scholar
[33] Zólyomi V, Drummond N D, Fal'ko V I 2014 Phys. Rev. B 89 205416
Google Scholar
[34] Zheng H, Li X B, Chen N K, Xie S Y, Tian W Q, Chen Y, Xia H, Zhang S B, Sun H B 2015 Phys. Rev. B 92 115307
Google Scholar
[35] Monroy E, Omn s F, Calle F 2003 Semicond. Sci. Technol. 18 R33
Google Scholar
[36] Caraveo Frescas J A, Nayak P K, Al Jawhari H A, Granato D B, Schwingenschlögl U, Alshareef H N 2013 ACS Nano 7 5160
Google Scholar

 下载:
下载:
计量
- 文章访问数: 11112
- PDF下载量: 247
- 被引次数: 0
