










-
开发高效、稳定的电催化剂是燃料电池走向实用的关键. 为了解决催化剂因尺寸效应引起的催化活性和稳定性之间的矛盾, 采用简便的一步溶剂热法设计合成了具有一维链状结构的Pt-Ni合金纳米颗粒催化剂. 链状Pt-Ni纳米颗粒由平均尺寸约为10 nm的纳米颗粒和直径约为3 nm, 长度为几百纳米的纳米线组装而成, 该结构具有零维纳米颗粒高的比表面积和一维纳米线高的结构稳定性优势, 可显著提高甲醇氧化反应的催化活性和稳定性, 其质量活性和比活性分别是商业Pt/C纳米催化剂的5.7倍和7.6倍. 经1000圈循环伏安测试后, 该纳米材料仍保留91.2%的比活性, 远高于商业Pt/C的4.4%. 制备的一维链状结构很好地解决了纳米颗粒催化剂在反应中的团聚问题, 为获得同时具有较高催化活性和稳定性的Pt基纳米催化剂提供了新的途径, 有望实现大范围工业化应用.
-
关键词:
- 链状Pt-Ni纳米颗粒 /
- 一维组装纳米结构 /
- 直接甲醇燃料电池
Fuel cells are one of the promising energy-conversion devices due to their high efficiency and zero emission. Despite tremendous research works in past decades, there remains a tough challenge in realizing the commercial applications of fuel cell technologies. Therefore, the development of highly efficient and stable fuel cell electrocatalyst is the top priority for practical fuel cells. As we all know, the small-size nanoparticles always have high specific surface area, which can provide more active sites to enhance the catalytic activity, while the one-dimensional nanowires usually own high structural stability. It may provide a possibility for the design of a novel bimetal Pt-based alloy nanostructure by combining the structural superiority of both, which can maintain the high stability and maximize the catalytic activity at the same time. Driven by these purposes, a novel nanostructure constructed by Pt-Ni alloy nanoparticles with a one-dimensional chain structure was designed to balance the contradiction between the activity and stability due to the size effects (the smaller the size, the higher the activity, and the worse the stability of the nanocatalyst; and vice versa). Here, a simple one-step solvothermal method has been adopted to produce the novel nanostructures constructed by the chain-like Pt-Ni nanoparticles (Pt-Ni CNPs) with Pt-rich crystal faces and alloy nature. The structure, component and catalysis were investigated by the combination of X-ray diffraction, transmission electron microscopy, X-ray photoemission spectra, and electrochemical measurements. The results show that the as-synthesized Pt-Ni CNP is constructed from a nanowire (with a diameter of about 3 nm and a length of several hundred nanometers) and the nanoparticles (with an average diameter of about 10 nm). This nanostructure is cleverly integrated the structural advantages of one-dimensional nanowires and zero-dimensional nanoparticles, which can significantly enhance the catalytic activity and stability for the methanol oxidation reaction (MOR) in acidic environment. Specially, the mass activity and specific activity of as-prepared Pt-Ni CNPs are 5.7 and 7.6 times higher than those of the commercial Pt/C, respectively. After 1000 cycles of cyclic voltammetry (CV) measurement, Pt-Ni CNPs still retain 91.2% of the specific activity, while the commercial Pt/C undergoes a drastic loss of MOR activities, retaining only 4.4% of the initial activity. It is particularly noteworthy that this nanostructure of Pt-Ni CNP solves the problem of agglomeration of nanoparticle catalysts in the reaction, and provides a new approach to obtain Pt-based nanocatalysts with high catalytic activity and stability at the same time. Our finding will provide insight into more rational designs of Pt-based bimetallic nanocatalysts with one-dimensional architectures, which is expected to promote the further development and large-scale industrial application of the direct methanol fuel.-
Keywords:
- chain-like PtNi nanoparticles /
- one-dimensional assembly nanostructure /
- direct methanol fuel
[1] Guo S J, Zhang S, Sun S H 2013 Angew. Chem. Int. Ed. 52 8526
 Google Scholar
Google Scholar
[2] Yan Z X, Xie J M, Shen P K 2015 J. Power Sources 286 239
Google Scholar
[3] 陈熙, 林正喆, 殷聪, 汤浩, 胡蕴成, 宁西京 2012 物理学报 61 076801
Google Scholar
Chen X, Lin Z Z, Yin C, Tang H, Hu Y C, Ning X J 2012 Acta. Phys. Sin. 61 076801
Google Scholar
[4] Cui Z M, Chen H, Zhao M T, Marshall D, Yu Y C, Abruna H, DiSalvo F J 2014 J. Am. Chem. Soc. 136 10206
Google Scholar
[5] Vandichel M, Moscu A, Grönbeck H 2017 ACS Catal. 7 7431
Google Scholar
[6] Guo S J, Wen D, Zhai Y M, Dong S J, Wang E 2010 ACS Nano 4 3959
Google Scholar
[7] Yan Y C, Shan H, Li G, Xiao F, Jiang Y Y, Yan Y Y, Jin C H, Zhang H, Wu J B, Yang D R 2016 Nano Lett. 16 7999
Google Scholar
[8] 田惠忱, 刘丽, 文玉华 2009 物理学报 58 4080
Google Scholar
Tian H C, Liu L, Wen Y H 2009 Acta. Phys. Sin. 58 4080
Google Scholar
[9] Chen C, Kang Y J, Huo Z Y, Zhu Z W, Huang W Y, Xin H L L, Snyder J D, Li D G, Herron J A, Mavrikakis M 2014 Science 343 1339
Google Scholar
[10] Greeley J, Stephens I E L, Bondarenko A S, Johansson T P, Hansen H A, Jaramillo T F, Rossmeisl J, Chorkendorff I, Nørskov J K 2009 Nat. Chem. 1 552
Google Scholar
[11] Bu L Z, Zhang N, Guo S J, Zhang X, Li J, Yao J L, Wu T, Lu G, Ma J Y, Su D 2016 Science 354 1410
Google Scholar
[12] Strasser P, Koh S, Anniyev T, Greeley J, More K, Yu C F, Liu Z C, Kaya S, Nordlund D, Ogasawara H, Toney M F, Nilsson A 2010 Nat. Chem. 2 454
Google Scholar
[13] 汪志刚, 黄娆, 文玉华 2013 物理学报 62 126101
Google Scholar
Wang Z G, Huang R, Wen Y H 2013 Acta. Phys. Sin. 62 126101
Google Scholar
[14] Wang D S, Zhao P, Li Y D 2011 Sci. Rep.-UK 1 37
Google Scholar
[15] Gu J, Zhang Y W, Tao F F 2012 Chem. Soc. Rev. 41 8050
Google Scholar
[16] 孙世刚, 文玉华, 张杨, 朱梓忠 2009 物理学报 58 2585
Google Scholar
Sun S G, Wen Y H, Zhang Y, Zhu Z Z 2009 Acta. Phys. Sin. 58 2585
Google Scholar
[17] Zhou X W, Zhang R H, Zhou Z Y, Sun S G 2011 J. Power Sources 196 5844
Google Scholar
[18] Shan A X, Chen Z C, Li B Q, Chen C P, Wang R M 2015 J. Mater. Chem. A 3 1031
Google Scholar
[19] Zhang L W, Gao A, Liu Y, Wang Y, Ma J T 2014 Electrochim. Acta 132 416
Google Scholar
[20] Chung D Y, Yoo J M, Sung Y E 2018 Adv. Mater. 30 1704123
Google Scholar
[21] Kong D S, Cha J J, Wang H T, Lee H R, Cui Y 2013 Energy Environ. Sci. 6 3553
Google Scholar
[22] Xia B Y, Wu H B, Li N, Yan Y, Lou X W, Wang X 2015 Angew. Chem. Int. Ed. 54 3797
Google Scholar
[23] Yoo S H, Park S 2007 Adv. Mater. 19 1612
Google Scholar
[24] Liu F, Lee J Y, Zhou W J 2006 Small 2 121
Google Scholar
[25] Kim J M, Joh H I, Jo S M, Ahn D J, Ha H Y, Hong S A, Kim S K 2010 Electrochim. Acta 55 4827
Google Scholar
[26] Liang H W, Cao X, Zhou F, Cui C H, Zhang W J, Yu S H 2011 Adv. Mater. 23 1467
Google Scholar
[27] Ding L X, Li G R, Wang Z L, Liu Z Q, Liu H, Tong Y X 2012 Chem. Eur. J. 18 8386
Google Scholar
[28] Guo S J, Zhang S, Sun X L, Sun S H 2011 J. Am. Chem. Soc. 133 15354
Google Scholar
[29] Tian X L, Zhao X, Su Y Q, Wang L J, Wang H M, Dang D, Chi B, Liu H F, Hensen E J, Lou X W, Xia B Y 2019 Science 366 850
Google Scholar
[30] Luo M C, Sun Y J, Zhang X, Qin Y N, Li M Q, Li Y J, Li C J, Yang Y, Wang L, Gao P, Lu G, Guo S J 2018 Adv. Mater. 30 1705515
Google Scholar
[31] Gao F, Zhang Y P, Song P P, Wang J, Yan B, Sun Q W, Li L, Zhu X, Du Y K 2019 Nanoscale 11 4831
Google Scholar
[32] Bu L Z, Ding J B, Guo S J, Zhang X, Su D, Zhu X, Yao J L, Guo J, Lu G, Huang X Q 2015 Adv. Mater. 27 7204
Google Scholar
[33] Zhao Y P, Tao L, Dang W, Wang L L, Xia M R, Wang B, Liu M M, Gao F M, Zhang J J, Zhao Y F 2019 Small 15 1900288
Google Scholar
[34] Qiu X Y, Li T C, Deng S H, Cen K, Xu L D, Tang Y W 2018 Chem. Eur. J. 24 1246
Google Scholar
[35] Tseng Y C, Chen H S, Liu C W, Yeh T H, Wang K W 2014 J. Mater. Chem. A 2 4270
Google Scholar
[36] Wagner C D 1979 Perkin-Elmer Corporation 80
[37] Wang J, Yang B B, Gao F, Song P P, Li L, Zhang Y P, Lu C, Goh M C, Du Y K 2019 Nanoscale Res. Lett. 14 11
Google Scholar
[38] Zhao X, Zhang J, Wang L J, Li H X, Liu Z L, Chen W 2015 ACS Appl. Mater. Interfaces 7 26333
Google Scholar
[39] Liu H M, Liu X Y, Li Y M, Jia Y J, Tang Y W, Chen Y 2016 Nano Res. 9 3494
Google Scholar
[40] Xie L S, Liu Q, Shi X F, Asiri A M, Luo Y L, Sun X P 2018 Inorg. Chem. Front. 5 1365
Google Scholar
[41] Van der Vliet D F, Wang C, Li D G, Paulikas A P, Greeley J, Rankin R B, Strmcnik D, Tripkovic D, Markovic N M, Stamenkovic V R 2012 Angew. Chem. Int. Ed. 51 3139
Google Scholar
[42] Xia T Y, Liu J L, Wang S G, Wang C, Sun Y, Gu L, Wang R M 2016 ACS Appl. Mater. Interfaces 8 10841
Google Scholar
[43] Li K, Li X X, Huang H W, Luo L H, Li X, Yan X P, Ma C, Si R, Yang J L, Zeng J 2018 J. Am. Chem. Soc. 140 16159
Google Scholar
[44] Debe M K 2012 Nature 486 43
Google Scholar
[45] Wang D Y, Chou H L, Lin Y C, Lai F J, Chen C H, Lee J F, Hwang B J, Chen C C 2012 J. Am. Chem. Soc. 134 10011
Google Scholar
[46] Liu H Q, Adzic R R, Wong S S 2015 ACS Appl. Mater. Interfaces 7 26145
Google Scholar
[47] Lai S Q, Fu C L, Chen Y X, Yu X, Lai X D, Ye C, Hu J Q 2015 J. Power Sources 274 604
Google Scholar
[48] Zhang X R, Fan H S, Zheng J L, Duan S B, Huang Y X, Cui Y M, Wang R M 2018 Catal. Sci. Technol. 8 4757
Google Scholar
[49] Du C Y, Chen M, Wang W G, Yin G P 2011 ACS Appl. Mater. Interfaces 3 105
Google Scholar
[50] Xia T Y, Liu J L, Wang S G, Wang C, Sun Y, Wang R M 2016 Sci. China Mater. 60 57
Google Scholar
[51] Lim K H, Chen Z X, Neyman K M, Rösch N 2006 J. Phys. Chem. B 110 14890
Google Scholar
[52] Li M F, Zhao Z P, Cheng T, Fortunelli A, Chen C Y, Yu R, Zhang Q H, Gu L, Merinov B V, Lin Z Y 2016 Science 354 1414
Google Scholar
[53] Zhang S, Zhang X, Jiang G M, Zhu H Y, Guo S Y, Su D, Lu G, Sun S H 2014 J. Am. Chem. Soc. 136 7734
Google Scholar
[54] Wang D L, Xin H L, Hovden R, Wang H, Yu Y C, Muller D A, DiSalvo F J, Abruña H D 2013 Nat. Mater. 12 81
Google Scholar
-

图 1 (a) Pt-Ni CNPs的TEM图像, 插图是纳米颗粒的直径分布统计图; (b) 高放大倍数下单根Pt-Ni CNPs的TEM图像; (c) Pt-Ni CNPs的HAADF-STEM图像; (d)和(e)分别为(c)中Pt和Ni的EDS元素分布
Fig. 1. (a) TEM image of Pt-Ni CNPs. Inset: graph of the diameter distribution of nanoparticles; (b) TEM image of a single Pt-Ni CNPs at a higher magnification; (c) HAADF-STEM images of Pt-Ni CNPs; (d) and (e) are EDS element distribution images of Pt and Ni in Pt-Ni CNPs corresponding to (c), respectively.
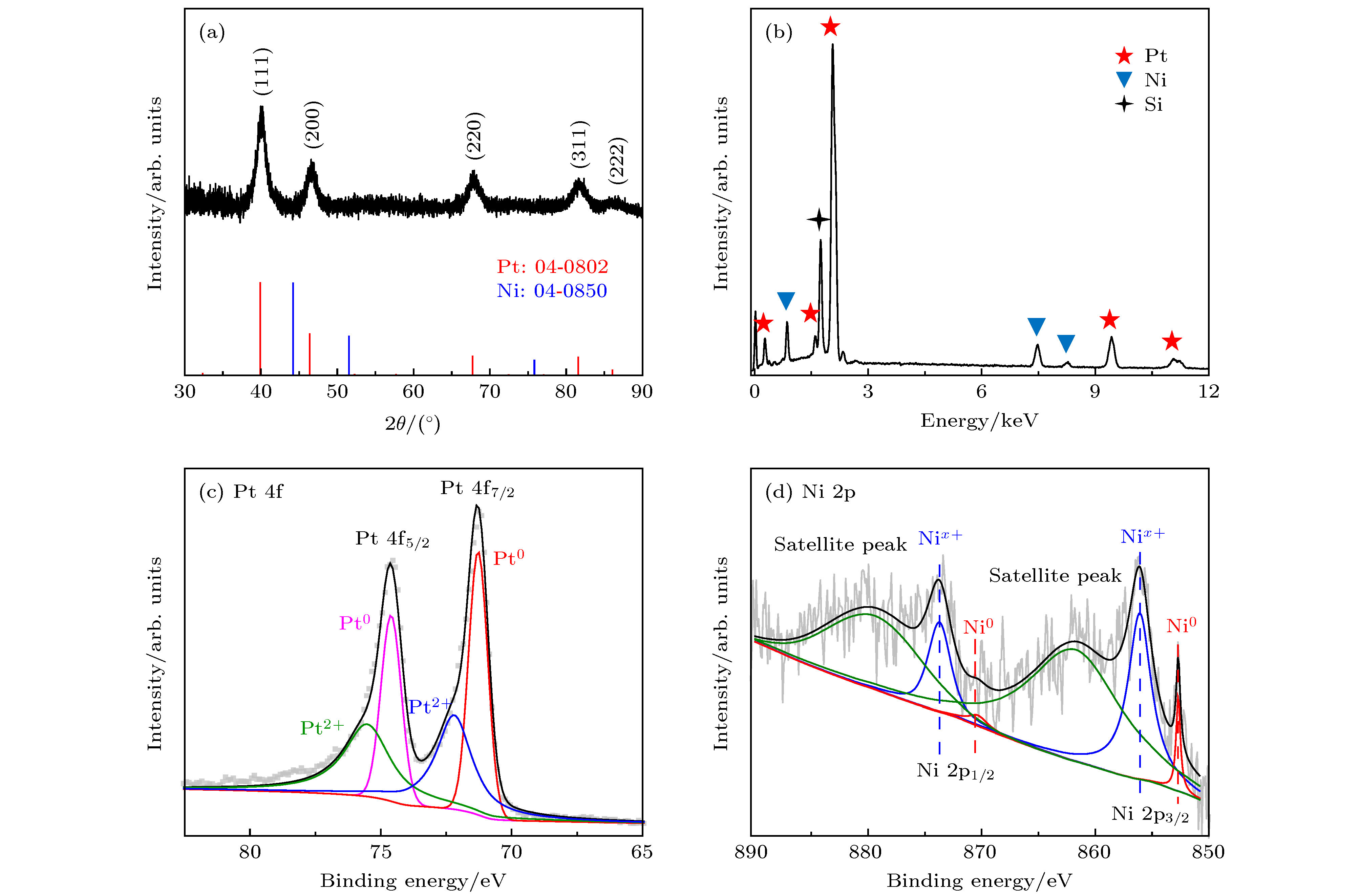
图 2 (a) Pt-Ni CNPs的 XRD 谱图及Pt 和 Ni 的标准卡片峰(分别对应红色和蓝色); (b) Pt-Ni CNPs的 EDS能谱图; (c)和(d)为Pt-Ni CNPs的XPS谱图, 分别对应Pt的4f峰和Ni的2p峰
Fig. 2. (a) XRD patterns of Pt-Ni CNPs and standard card peaks of Pt and Ni (corresponding to red and blue respectively); (b) EDS spectrum of Pt-Ni CNPs; (c) and (d) are XPS spectra of Pt-Ni CNPs, corresponding to the 4f peak of Pt and the 2p peak of Ni, respectively.
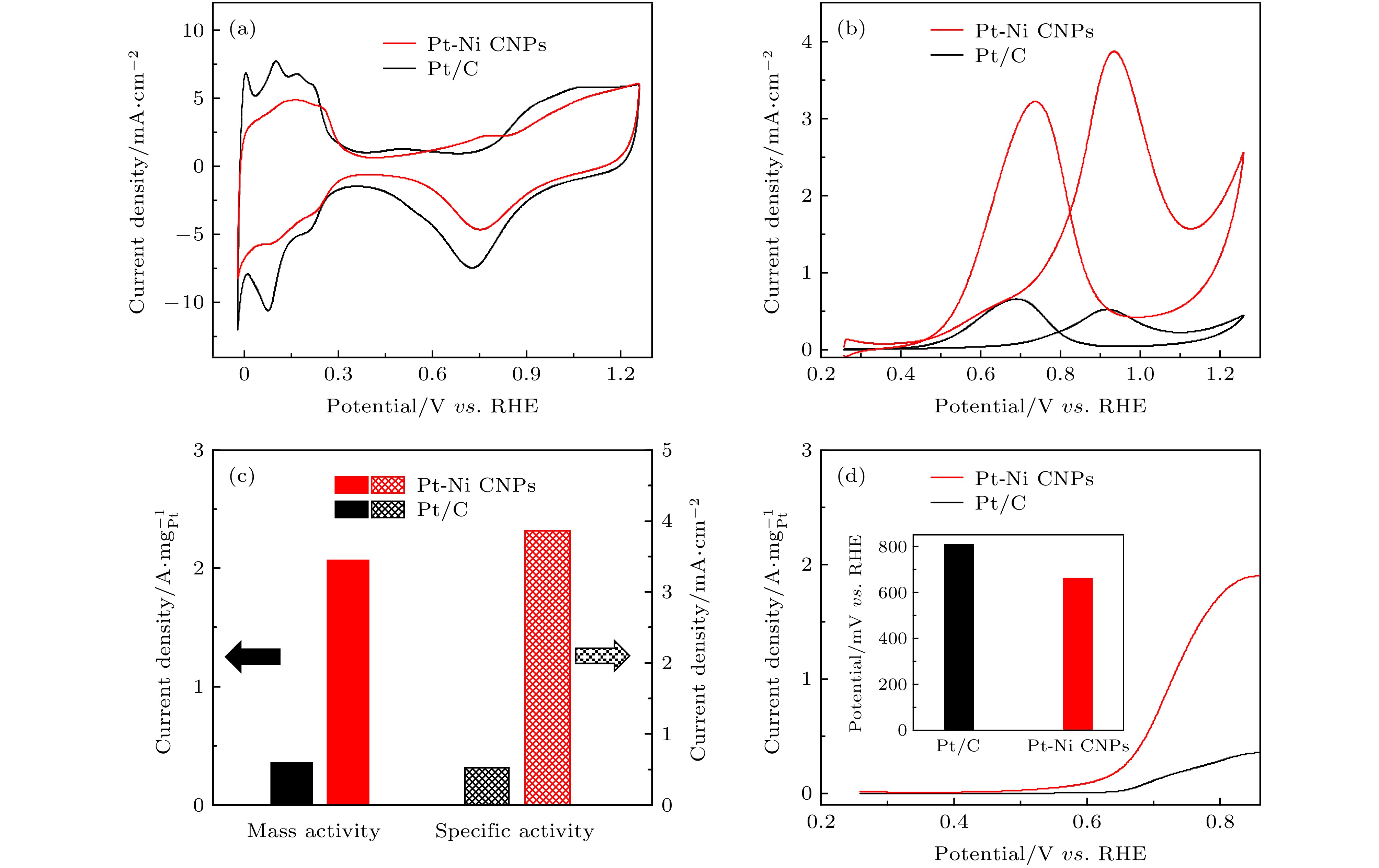
图 3 Pt-Ni CNPs (红色) 和商业Pt/C (黑色) 的MOR性能对比 (a), (b)两种催化剂的CV曲线, 分别是ECSA和MOR; (c) 两种样品相对应的质量活性和比活性; (d) 以5 mV/s的扫描速率测得的LSV曲线, 插图是固定电流密度所需提供的电位值
Fig. 3. MOR performance comparison for Pt-Ni CNPs (red) and commercial Pt/C (black): (a) CV of the above catalysts for ECSAs; (b) CV of the above catalysts for MOR; (c) corresponding mass and specific activities of different catalysts for MOR; (d) LSV curves of the above electrocatalysts with a low scan rate of 5 mV/s. Inset:the potential required for fixed current density.
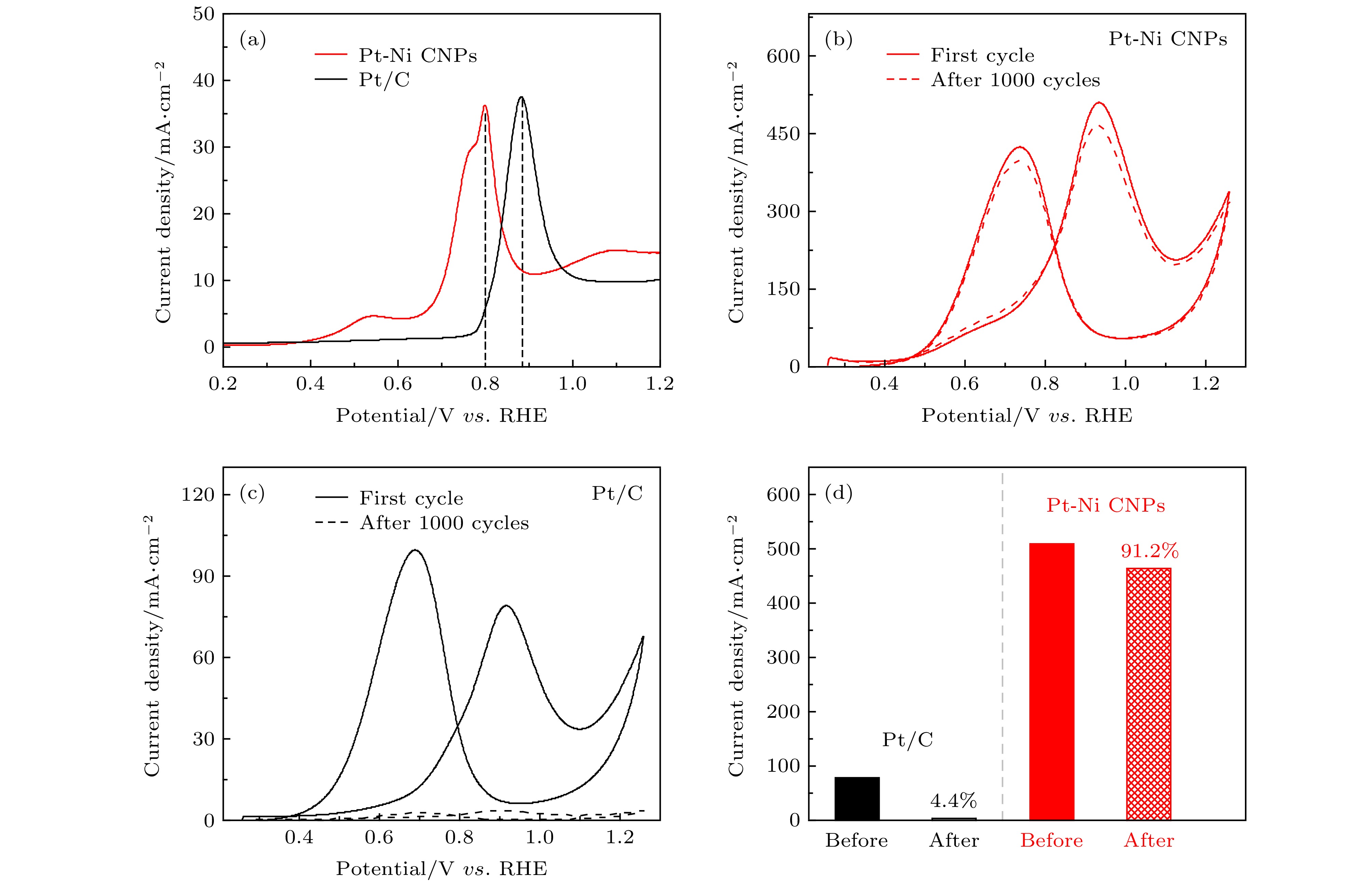
图 4 (a) CO溶解曲线; (b)和(c)分别为Pt-Ni CNPs (红色)和商业Pt/C (黑色)在0.5 M H2SO4和1 M CH3OH混合溶液中的稳定性测试: 实线为第一圈CV循环曲线, 虚线为第1000圈CV循环曲线; (d) 两种样品1000圈CV循环前后比活性对比
Fig. 4. (a) The electrode area-normalized CO stripping curves; Stability test in 0.5 M H2SO4 and 1 M CH3OH solutions: (b) Pt-Ni CNPs (red) and (c) commercial Pt/C (black) with solid line as the first cycle and dashed line as the 1000th cycle; (d) specific activities of two samples before and after 1000 cycles.
-
[1] Guo S J, Zhang S, Sun S H 2013 Angew. Chem. Int. Ed. 52 8526
Google Scholar
[2] Yan Z X, Xie J M, Shen P K 2015 J. Power Sources 286 239
Google Scholar
[3] 陈熙, 林正喆, 殷聪, 汤浩, 胡蕴成, 宁西京 2012 物理学报 61 076801
Google Scholar
Chen X, Lin Z Z, Yin C, Tang H, Hu Y C, Ning X J 2012 Acta. Phys. Sin. 61 076801
Google Scholar
[4] Cui Z M, Chen H, Zhao M T, Marshall D, Yu Y C, Abruna H, DiSalvo F J 2014 J. Am. Chem. Soc. 136 10206
Google Scholar
[5] Vandichel M, Moscu A, Grönbeck H 2017 ACS Catal. 7 7431
Google Scholar
[6] Guo S J, Wen D, Zhai Y M, Dong S J, Wang E 2010 ACS Nano 4 3959
Google Scholar
[7] Yan Y C, Shan H, Li G, Xiao F, Jiang Y Y, Yan Y Y, Jin C H, Zhang H, Wu J B, Yang D R 2016 Nano Lett. 16 7999
Google Scholar
[8] 田惠忱, 刘丽, 文玉华 2009 物理学报 58 4080
Google Scholar
Tian H C, Liu L, Wen Y H 2009 Acta. Phys. Sin. 58 4080
Google Scholar
[9] Chen C, Kang Y J, Huo Z Y, Zhu Z W, Huang W Y, Xin H L L, Snyder J D, Li D G, Herron J A, Mavrikakis M 2014 Science 343 1339
Google Scholar
[10] Greeley J, Stephens I E L, Bondarenko A S, Johansson T P, Hansen H A, Jaramillo T F, Rossmeisl J, Chorkendorff I, Nørskov J K 2009 Nat. Chem. 1 552
Google Scholar
[11] Bu L Z, Zhang N, Guo S J, Zhang X, Li J, Yao J L, Wu T, Lu G, Ma J Y, Su D 2016 Science 354 1410
Google Scholar
[12] Strasser P, Koh S, Anniyev T, Greeley J, More K, Yu C F, Liu Z C, Kaya S, Nordlund D, Ogasawara H, Toney M F, Nilsson A 2010 Nat. Chem. 2 454
Google Scholar
[13] 汪志刚, 黄娆, 文玉华 2013 物理学报 62 126101
Google Scholar
Wang Z G, Huang R, Wen Y H 2013 Acta. Phys. Sin. 62 126101
Google Scholar
[14] Wang D S, Zhao P, Li Y D 2011 Sci. Rep.-UK 1 37
Google Scholar
[15] Gu J, Zhang Y W, Tao F F 2012 Chem. Soc. Rev. 41 8050
Google Scholar
[16] 孙世刚, 文玉华, 张杨, 朱梓忠 2009 物理学报 58 2585
Google Scholar
Sun S G, Wen Y H, Zhang Y, Zhu Z Z 2009 Acta. Phys. Sin. 58 2585
Google Scholar
[17] Zhou X W, Zhang R H, Zhou Z Y, Sun S G 2011 J. Power Sources 196 5844
Google Scholar
[18] Shan A X, Chen Z C, Li B Q, Chen C P, Wang R M 2015 J. Mater. Chem. A 3 1031
Google Scholar
[19] Zhang L W, Gao A, Liu Y, Wang Y, Ma J T 2014 Electrochim. Acta 132 416
Google Scholar
[20] Chung D Y, Yoo J M, Sung Y E 2018 Adv. Mater. 30 1704123
Google Scholar
[21] Kong D S, Cha J J, Wang H T, Lee H R, Cui Y 2013 Energy Environ. Sci. 6 3553
Google Scholar
[22] Xia B Y, Wu H B, Li N, Yan Y, Lou X W, Wang X 2015 Angew. Chem. Int. Ed. 54 3797
Google Scholar
[23] Yoo S H, Park S 2007 Adv. Mater. 19 1612
Google Scholar
[24] Liu F, Lee J Y, Zhou W J 2006 Small 2 121
Google Scholar
[25] Kim J M, Joh H I, Jo S M, Ahn D J, Ha H Y, Hong S A, Kim S K 2010 Electrochim. Acta 55 4827
Google Scholar
[26] Liang H W, Cao X, Zhou F, Cui C H, Zhang W J, Yu S H 2011 Adv. Mater. 23 1467
Google Scholar
[27] Ding L X, Li G R, Wang Z L, Liu Z Q, Liu H, Tong Y X 2012 Chem. Eur. J. 18 8386
Google Scholar
[28] Guo S J, Zhang S, Sun X L, Sun S H 2011 J. Am. Chem. Soc. 133 15354
Google Scholar
[29] Tian X L, Zhao X, Su Y Q, Wang L J, Wang H M, Dang D, Chi B, Liu H F, Hensen E J, Lou X W, Xia B Y 2019 Science 366 850
Google Scholar
[30] Luo M C, Sun Y J, Zhang X, Qin Y N, Li M Q, Li Y J, Li C J, Yang Y, Wang L, Gao P, Lu G, Guo S J 2018 Adv. Mater. 30 1705515
Google Scholar
[31] Gao F, Zhang Y P, Song P P, Wang J, Yan B, Sun Q W, Li L, Zhu X, Du Y K 2019 Nanoscale 11 4831
Google Scholar
[32] Bu L Z, Ding J B, Guo S J, Zhang X, Su D, Zhu X, Yao J L, Guo J, Lu G, Huang X Q 2015 Adv. Mater. 27 7204
Google Scholar
[33] Zhao Y P, Tao L, Dang W, Wang L L, Xia M R, Wang B, Liu M M, Gao F M, Zhang J J, Zhao Y F 2019 Small 15 1900288
Google Scholar
[34] Qiu X Y, Li T C, Deng S H, Cen K, Xu L D, Tang Y W 2018 Chem. Eur. J. 24 1246
Google Scholar
[35] Tseng Y C, Chen H S, Liu C W, Yeh T H, Wang K W 2014 J. Mater. Chem. A 2 4270
Google Scholar
[36] Wagner C D 1979 Perkin-Elmer Corporation 80
[37] Wang J, Yang B B, Gao F, Song P P, Li L, Zhang Y P, Lu C, Goh M C, Du Y K 2019 Nanoscale Res. Lett. 14 11
Google Scholar
[38] Zhao X, Zhang J, Wang L J, Li H X, Liu Z L, Chen W 2015 ACS Appl. Mater. Interfaces 7 26333
Google Scholar
[39] Liu H M, Liu X Y, Li Y M, Jia Y J, Tang Y W, Chen Y 2016 Nano Res. 9 3494
Google Scholar
[40] Xie L S, Liu Q, Shi X F, Asiri A M, Luo Y L, Sun X P 2018 Inorg. Chem. Front. 5 1365
Google Scholar
[41] Van der Vliet D F, Wang C, Li D G, Paulikas A P, Greeley J, Rankin R B, Strmcnik D, Tripkovic D, Markovic N M, Stamenkovic V R 2012 Angew. Chem. Int. Ed. 51 3139
Google Scholar
[42] Xia T Y, Liu J L, Wang S G, Wang C, Sun Y, Gu L, Wang R M 2016 ACS Appl. Mater. Interfaces 8 10841
Google Scholar
[43] Li K, Li X X, Huang H W, Luo L H, Li X, Yan X P, Ma C, Si R, Yang J L, Zeng J 2018 J. Am. Chem. Soc. 140 16159
Google Scholar
[44] Debe M K 2012 Nature 486 43
Google Scholar
[45] Wang D Y, Chou H L, Lin Y C, Lai F J, Chen C H, Lee J F, Hwang B J, Chen C C 2012 J. Am. Chem. Soc. 134 10011
Google Scholar
[46] Liu H Q, Adzic R R, Wong S S 2015 ACS Appl. Mater. Interfaces 7 26145
Google Scholar
[47] Lai S Q, Fu C L, Chen Y X, Yu X, Lai X D, Ye C, Hu J Q 2015 J. Power Sources 274 604
Google Scholar
[48] Zhang X R, Fan H S, Zheng J L, Duan S B, Huang Y X, Cui Y M, Wang R M 2018 Catal. Sci. Technol. 8 4757
Google Scholar
[49] Du C Y, Chen M, Wang W G, Yin G P 2011 ACS Appl. Mater. Interfaces 3 105
Google Scholar
[50] Xia T Y, Liu J L, Wang S G, Wang C, Sun Y, Wang R M 2016 Sci. China Mater. 60 57
Google Scholar
[51] Lim K H, Chen Z X, Neyman K M, Rösch N 2006 J. Phys. Chem. B 110 14890
Google Scholar
[52] Li M F, Zhao Z P, Cheng T, Fortunelli A, Chen C Y, Yu R, Zhang Q H, Gu L, Merinov B V, Lin Z Y 2016 Science 354 1414
Google Scholar
[53] Zhang S, Zhang X, Jiang G M, Zhu H Y, Guo S Y, Su D, Lu G, Sun S H 2014 J. Am. Chem. Soc. 136 7734
Google Scholar
[54] Wang D L, Xin H L, Hovden R, Wang H, Yu Y C, Muller D A, DiSalvo F J, Abruña H D 2013 Nat. Mater. 12 81
Google Scholar

 下载:
下载:
计量
- 文章访问数: 26386
- PDF下载量: 412
- 被引次数: 0
