










-
用氢对单层二维过渡金属硫化物(TMDs)进行功能化是调节单层TMDs电子性质的既有效又经济的方法. 采用密度泛函理论, 对单层TMDs (MX2 (M = Mo, W; X = S, Se, Te))的稳定性和电子性质进行理论研究, 发现在单层MX2的层间有一个比其表面更稳定的氢吸附位点. 当同阳离子时, 随着阴离子原子序数的增加, H原子与MX2层的结合越强, 氢化单层MX2结构越稳定; 相反, 同阴离子时, 随着阳离子原子序数的增加, H原子与MX2层的结合越弱. 氢原子从MoS2的表面经层间穿越到另一表面的扩散势垒约为0.9 eV. 氢化对单层MX2的电子特性也会产生极大的影响, 主要表现在氢化实现了MX2体系从无磁性到磁性体系的过渡. 表面氢化会使MX2层的带隙急剧减小, 而层间氢化使MX2的电子结构从半导体转变为金属能带.Chemical functionalization of two-dimensional transition metal dichalcogenides (TMDs) with hydrogen is an effective and economical method to synthesize monolayer TMDs and tune their electronic properties. We theoretically study the stabilities and electronic properties of chemisorbed H atoms on monolayer TMDs by using density-functional theory calculations. The result shows that there exists a more stable adsorption site in the layers of the monolayer MX2 (M = Mo, W; X = S, Se, Te) than its surface for hydrogen. In the case of the same cation, with the increase of the anion (X2−) atomic number, the stronger the bonding between the H atom and the MX2 layer, the more stable the structure of the hydrogenated monolayer MX2 is. However, in the case of the same anion, the binding between the H atom and the MX2 layer becomes weaker as the atomic number of the cations increases. H atoms passes through one surface of the MS2 to the other surface with a relatively small diffusion barrier of about 0.9 eV. So the H atoms can more easily go through the barrier. And for the H atom to go through the other monolayer MX2 (M = Mo, W; X = Se, Te), the diffusion barrier is about 1.2 eV. H atoms are difficult to pass through the barrier at this time. The singular diffusion behavior of H atoms in monolayer MX2 is conducible to understanding the stability of hydrogenated two-dimensional transition metal sulfide system. In addition, the surface hydrogenation and interlaminar hydrogenation have different effects on the electronic properties of monolayer MX2, and mainly manifest themselves in the fact that the surface hydrogenation induces spontaneous magnetism and sharply reduces the band gap, but still retains the semiconductor properties of the original monolayer MX2. However, interlaminar hydrogenation enables monolayer MX2 to directly realize the transition from semiconductor to metal. Interlaminar hydrogenation monolayer MX2 (M = Mo, W; X = S, Se) make the system generating magnetism, while when the anion is Te2−, the magnetism almost disappears. These results can provide theoretical guidance in understanding hydrogen functionalization of MX2 layer, and also present a certain theoretical basis for realizing the application of MX2 in nano-electronic devices.
-
Keywords:
- hydrogenation /
- two-dimensional transition metal dichalcogenides /
- stability /
- electronic properties
[1] Tang L P, Tang L M, Wang D, Deng H X, Chen K Q 2018 J. Phys.: Condens. Matter 30 465301
 Google Scholar
Google Scholar
[2] Xie G F, Ding D, Zhang G 2018 Adv. Phys. X 3 1480417
Google Scholar
[3] Wang D, Tang L M, Jiang X X, Tan J Y, He M D, Wang X J, Chen K Q 2018 Adv. Electron. Mater. DOI: 10.1002/aelm.201800475
[4] Koppens F H, Mueller T, Avouris P, Ferrari A C, Vitiello M S, Polini M 2014 Nat. Nanotechnol. 9 780
Google Scholar
[5] Schwierz F, Pezoldt J, Granzner R 2015 Nanoscale 7 8261
Google Scholar
[6] Dean C R, Young A F, Meric I, Lee C, Wang L, Sorgenfrei S, Watanabe K, Taniguchi T, Kim P, Shepard K L, Hone J 2010 Nat. Nanotechnol. 5 722
Google Scholar
[7] Zhang Y, Tang T T, Girit C, Hao Z, Martin M C, Zettl A, Crommie M F, Shen Y R, Wang F 2009 Nature 459 820
Google Scholar
[8] Zhang W, Lin C T, Liu K K, Tite T, Su C Y, Chang C H, Lee Y H, Chu C W, Wei K H, Kuo J L, Li L J 2011 ACS Nano 5 7517
Google Scholar
[9] Flores M Z, Autreto P A, Legoas S B, Galvao D S 2009 Nanotechnology 20 465704
Google Scholar
[10] Sofo J O, Chaudhari A S, Barber G D 2007 Phys. Rev. B 75 153401
Google Scholar
[11] Wang H, Yu L, Lee Y H, Shi Y, Hsu A, Chin M, Li L J, Dubey M, Kong J, Palacios T 2012 Nano Lett. 12 4674
Google Scholar
[12] Radisavljevic B, Radenovic A, Brivio J, Giacometti V, Kis A 2011 Nat. Nanotechnol. 6 147
Google Scholar
[13] Kang K, Xie S, Huang L, Han Y, Huang P Y, Mark K F, Kim C J, Muller D, Park J 2015 Nature 520 656
Google Scholar
[14] Tang L P, Tang L M, Geng H, Yi Y P, Wei Z, Chen K Q, Deng H X 2018 Appl. Phys. Lett. 112 012101
Google Scholar
[15] Xue X X, Feng Y X, Liao L, Chen Q J, Wang D, Tang L M, Chen K Q 2018 J. Phys.: Condens. Matter 30 125001
Google Scholar
[16] 颜送灵, 唐黎明, 赵宇清 2016 物理学报 65 077301
Google Scholar
Yan S L, Tang L M, Zhao Y Q 2016 Acta Phys. Sin. 65 077301
Google Scholar
[17] 李立明, 宁锋, 唐黎明 2015 物理学报 64 227303
Google Scholar
Li L M, Ning F, Tang L M 2015 Acta Phys. Sin. 64 227303
Google Scholar
[18] Bernardi M, Palummo M, Grossman J C 2013 Nano Lett. 13 3664
Google Scholar
[19] Lopezsanchez O, Lembke D, Kayci M, Radenovic A, Kis A 2013 Nat. Nanotechnol. 8 497
Google Scholar
[20] Bertolazzi S, Brivio J, Kis A 2011 ACS Nano 5 9703
Google Scholar
[21] Eda G, Yamaguchi H, Voiry D, Fujita T, Chen M, Chhowalla M 2011 Nano Lett. 11 5111
Google Scholar
[22] Ning F, Wang D, Feng Y X, Tang L M, Zhang Y, Chen K Q 2017 J. Mater. Chem. C 5 9429
Google Scholar
[23] Cao T, Wang G, Han W, Ye H, Zhu C, Shi J, Niu Q, Tan P, Wang E, Liu B, Feng J 2012 Nat. Commun. 3 887
Google Scholar
[24] Li Q, Tang L, Zhang C, Wang D, Chen Q J, Feng Y X, Tang L M, Chen K Q 2017 Appl. Phys. Lett. 111 171602
Google Scholar
[25] Xu Y, Li Y, Chen X, Zhang C, Zhang R, Lu P 2016 AIP Adv. 6 075001
Google Scholar
[26] Kam K K, Parkinson B A 1982 J. Phys. Chem. 86 463
Google Scholar
[27] Chen J, Li S L, Xu Q, Tanaka K 2002 Chem. Commun. 16 1722
Google Scholar
[28] Cheng F Y, Chen J, Gou X L 2006 Adv. Mater. 18 2561
Google Scholar
[29] Karunadasa H I, Montalvo E, Sun Y, Majda M, Long J R, Chang C J 2012 Science 335 698
Google Scholar
[30] Mouri S, Miyauchi Y, Matsuda K 2013 Nano Lett. 13 5944
Google Scholar
[31] Chhowalla M, Amaratunga G A 2000 Nature 407 164
Google Scholar
[32] Muratore C, Voevodin A A 2006 Surf. Coat. Technol. 201 4125
Google Scholar
[33] Stefanov M, Enyashin A N, Heine T, Seifert G 2008 J. Phys. Chem. C 112 17764
Google Scholar
[34] Mak K F, Lee C, Hone J, Shan J, Heinz T F 2010 Phys. Rev. Lett. 105 136805
Google Scholar
[35] Elias D C, Nair R R, Mohiuddin T M G, Morozov S V, Blake P, Halsall M P, Ferrari A C, Boukhvalov D W, Katsnelson M I, Geim A K, Novoselov K S 2009 Science 323 610
Google Scholar
[36] Zou J, Tang L M, Chen K Q, Feng Y X 2017 J. Phys.: Condens. Matter 30 065001
Google Scholar
[37] Zhang W, Zhang Z, Yang W 2015 J. Nanosci. Nanotechnol. 15 8075
Google Scholar
[38] van der Marel D, Molegraaf H J A, Zaanen J, Nussinov Z, Carbone F, Damascelli H, Eisaki H, Greven M, Kes P H, Li M 2003 Nature 425 271
Google Scholar
[39] Sundberg P, Moyes R B, Tomkinson J 1991 Bull. Soc. Chim. Belg. 100 967
Google Scholar
[40] Lozada-Hidalgo M, Zhang S, Hu S, Kravets V G, Rodriguez F J, Berdyugin A, Grigorenko A, Geim A K 2018 Nat. Nanotechnol. 13 300
Google Scholar
[41] Perdew J P, Burke K, Ernzerhof M 1996 Phys. Rev. Lett. 77 3865
Google Scholar
[42] Blöchl P E 1994 Phys. Rev. B 50 17953
Google Scholar
[43] Henkelman G, Uberuaga B P, Jónsson H 2000 J. Chem. Phys. 113 9901
Google Scholar
[44] Koh E W K, Chiu C H, LimY K, ZhangY W, Pan H 2012 Int. J. Hydrogen Energy 37 14323
Google Scholar
[45] Özçelik V O, Azadani J G, Yang C, Koester S J, Low T 2016 Phys. Rev. B 94 035125
Google Scholar
-
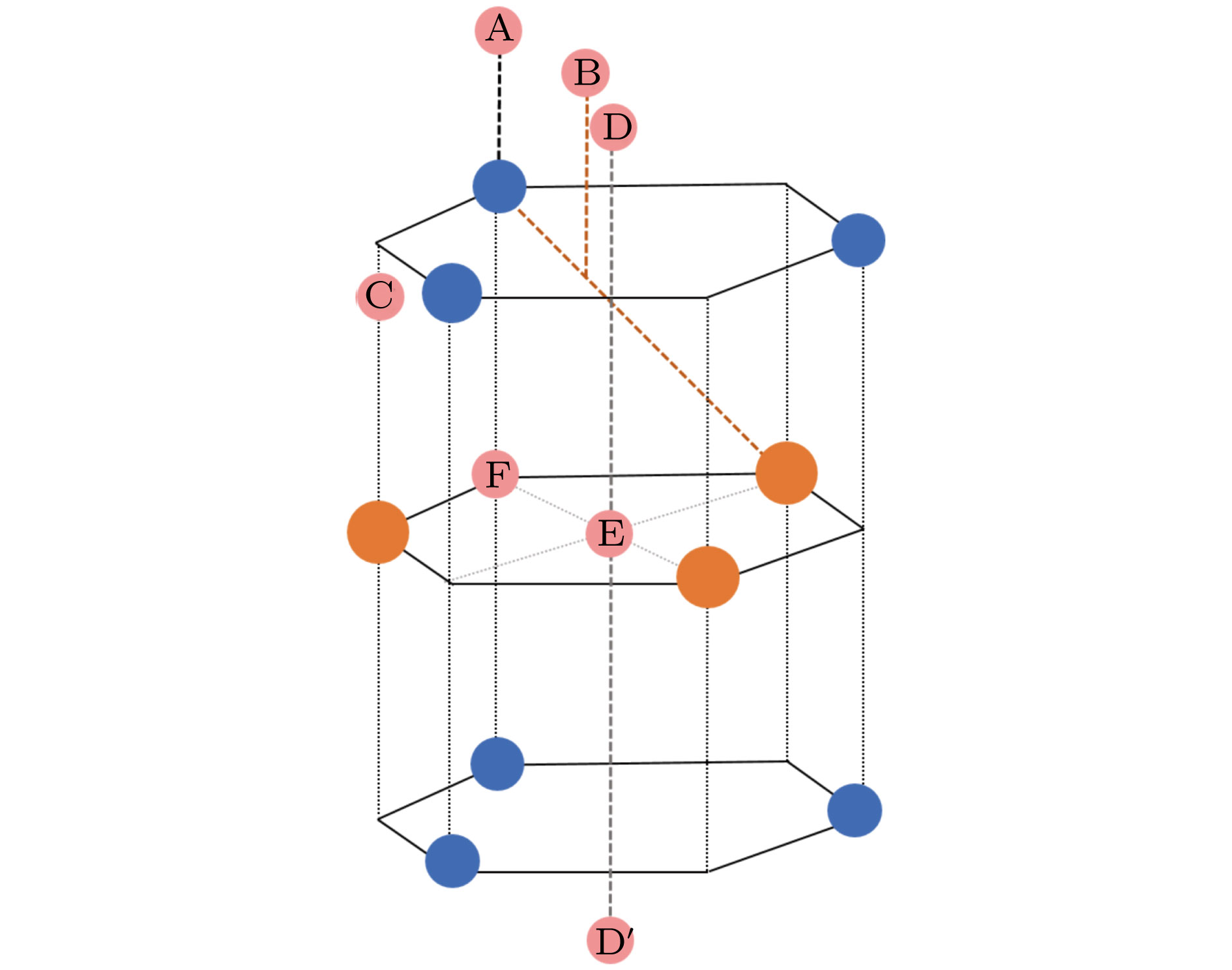
图 1 单层MX2的结构模型及可能的H吸附位点示意图, 其中蓝色, 橙色和粉色分别表示阴离子(X2−)、阳离子(M4+)和氢原子; A—F表示氢原子可能的吸附位点
Fig. 1. Structural model and possible H adsorption sites of single-layer MX2. Blue, orange and pink balls correspond to anions (X2−), cations (M4+) and hydrogen atoms, respectively. A−F shows a possible adsorption site for hydrogen atom.
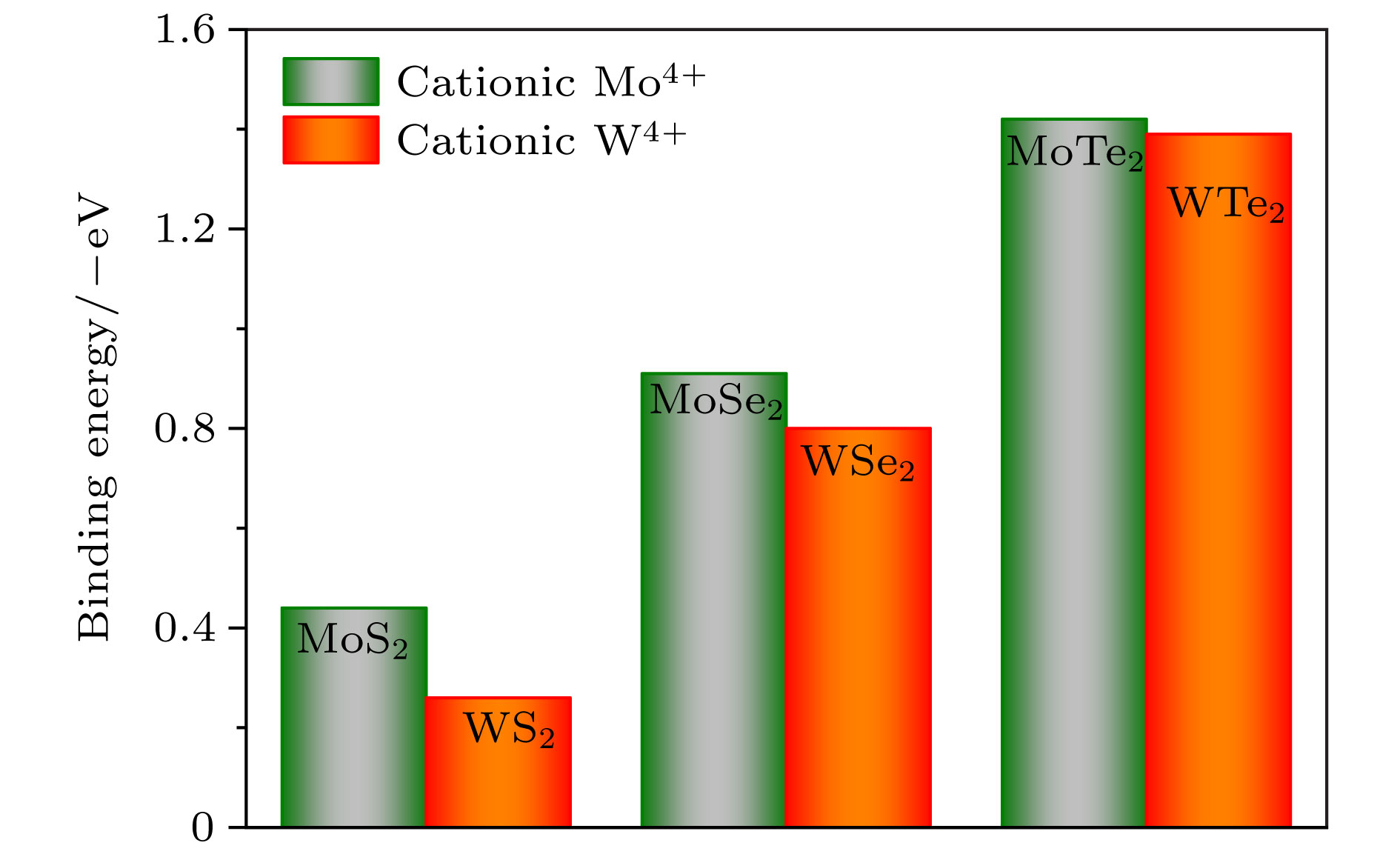
图 2 单层MX2的稳定吸附位点结合能统计图
Fig. 2. Binding energy of the stable adsorption sites of single-layer MX2.

图 3 氢原子穿过单层MX2的扩散势垒(扩散路径为从单层MX2表面D点穿过层间E点扩散到另一个表面D'点)
Fig. 3. Diffusion barrier of hydrogen atoms through a single layer of MX2. The diffusion path is from the D point of surface through the interlayer E point to the other surface D' point.
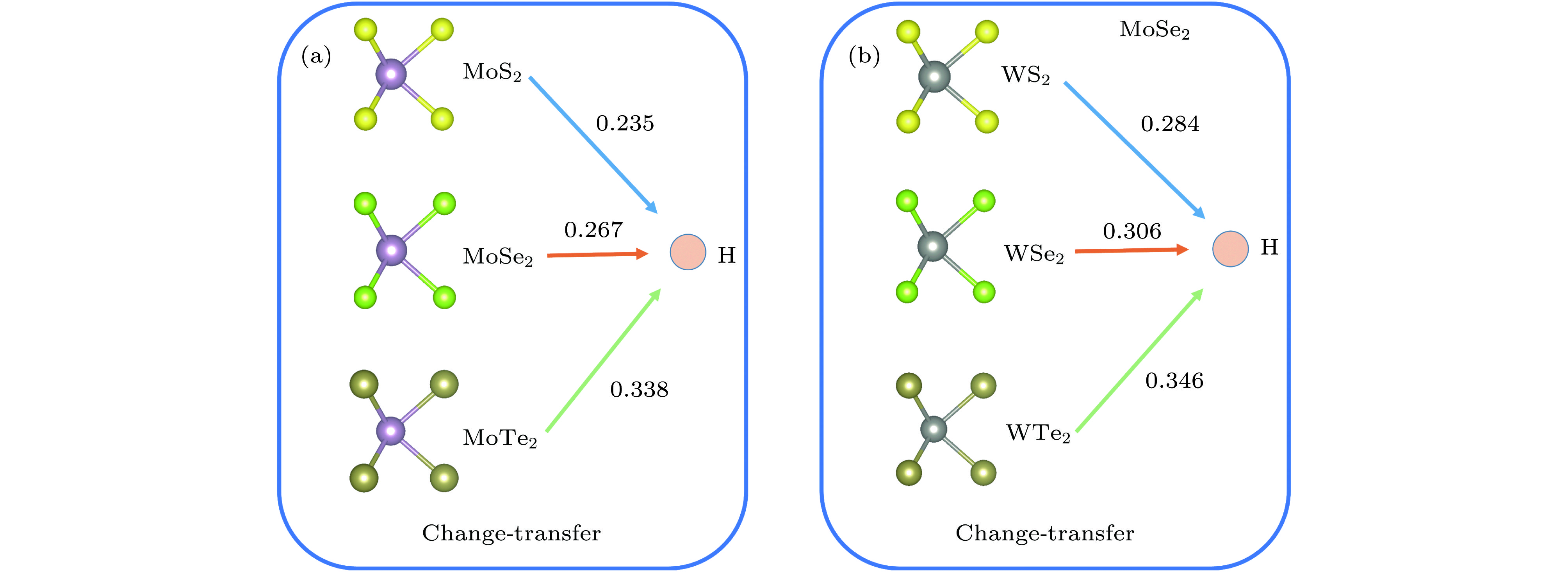
图 4 氢原子稳定吸附在单层 MX2的电荷转移分布图(计算的电荷转移为MX2层E位点的转移情况, 箭头指示电荷转移的方向)
Fig. 4. Charge transfer profile of hydrogen atoms adsorbed on a single-layer MX2. The calculated charge transfer is the transfer of the E site of the MX2 layer, and the arrows indicate the direction of charge transfer.
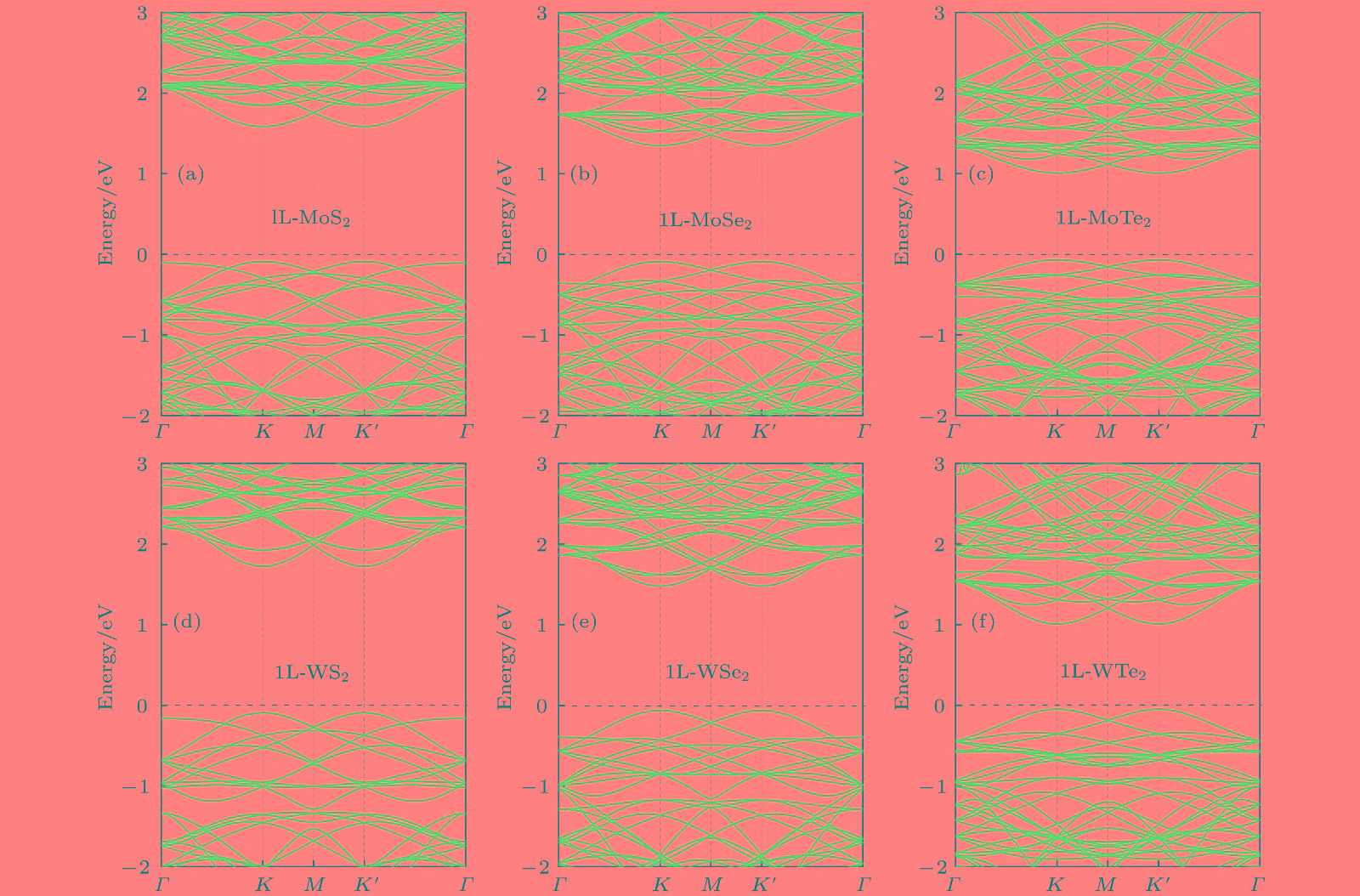
图 5 4 × 4单层MX2 (1L-MX2)超胞的能带结构图(费米能级用黑色虚线表述并设为0 eV)
Fig. 5. Band structures of 4 × 4 monolayer MX2 (1L-MX2) supercell. The fermi level is represented by dotted black line and set as 0 eV.

图 6 表面氢化MX2能带结构图(蓝色和红色线分别表示自旋向上和自旋向下)
Fig. 6. Band structures of surface hydrogenation MX2. Blue and red correspond to spin-up and spin-down, respectively.

图 7 层间氢化MX2能带结构图(蓝色和红色线表示自旋向上和自旋向下)
Fig. 7. Band structures of interlayer hydrogenation MX2. Blue and red correspond to spin-up and spin-down, respectively.
表 1 氢原子吸附在单层MX2上的可能吸附位点(AS)的结合能, 及氢原子与阳离子(M4+ = Mo, W)的键长
Table 1. Binding energy of possible adsorption sites (AS) of hydrogen atoms adsorbed on single-layer MX2, and M—H (M4+ = Mo, W) bond lengths.
AS Binding energy/eV M—H键长/Å A B C D E F MoS2 −0.38 −0.41 0.21 −0.27 −0.44 0.99 1.893 MoSe2 −0.19 −0.16 −0.26 −0.30 −0.91 0.47 1.950 MoTe2 −0.10 −0.35 −0.10 −0.52 −1.42 −0.19 1.996 WS2 −0.09 −0.16 0.30 −0.06 −0.26 1.58 1.901 WSe2 −0.01 −0.09 0.50 −0.18 −0.80 0.69 1.970 WTe2 0.09 −0.05 −0.64 −0.45 −1.39 −0.18 2.005  下载: 导出CSV
下载: 导出CSV
-
[1] Tang L P, Tang L M, Wang D, Deng H X, Chen K Q 2018 J. Phys.: Condens. Matter 30 465301
Google Scholar
[2] Xie G F, Ding D, Zhang G 2018 Adv. Phys. X 3 1480417
Google Scholar
[3] Wang D, Tang L M, Jiang X X, Tan J Y, He M D, Wang X J, Chen K Q 2018 Adv. Electron. Mater. DOI: 10.1002/aelm.201800475
[4] Koppens F H, Mueller T, Avouris P, Ferrari A C, Vitiello M S, Polini M 2014 Nat. Nanotechnol. 9 780
Google Scholar
[5] Schwierz F, Pezoldt J, Granzner R 2015 Nanoscale 7 8261
Google Scholar
[6] Dean C R, Young A F, Meric I, Lee C, Wang L, Sorgenfrei S, Watanabe K, Taniguchi T, Kim P, Shepard K L, Hone J 2010 Nat. Nanotechnol. 5 722
Google Scholar
[7] Zhang Y, Tang T T, Girit C, Hao Z, Martin M C, Zettl A, Crommie M F, Shen Y R, Wang F 2009 Nature 459 820
Google Scholar
[8] Zhang W, Lin C T, Liu K K, Tite T, Su C Y, Chang C H, Lee Y H, Chu C W, Wei K H, Kuo J L, Li L J 2011 ACS Nano 5 7517
Google Scholar
[9] Flores M Z, Autreto P A, Legoas S B, Galvao D S 2009 Nanotechnology 20 465704
Google Scholar
[10] Sofo J O, Chaudhari A S, Barber G D 2007 Phys. Rev. B 75 153401
Google Scholar
[11] Wang H, Yu L, Lee Y H, Shi Y, Hsu A, Chin M, Li L J, Dubey M, Kong J, Palacios T 2012 Nano Lett. 12 4674
Google Scholar
[12] Radisavljevic B, Radenovic A, Brivio J, Giacometti V, Kis A 2011 Nat. Nanotechnol. 6 147
Google Scholar
[13] Kang K, Xie S, Huang L, Han Y, Huang P Y, Mark K F, Kim C J, Muller D, Park J 2015 Nature 520 656
Google Scholar
[14] Tang L P, Tang L M, Geng H, Yi Y P, Wei Z, Chen K Q, Deng H X 2018 Appl. Phys. Lett. 112 012101
Google Scholar
[15] Xue X X, Feng Y X, Liao L, Chen Q J, Wang D, Tang L M, Chen K Q 2018 J. Phys.: Condens. Matter 30 125001
Google Scholar
[16] 颜送灵, 唐黎明, 赵宇清 2016 物理学报 65 077301
Google Scholar
Yan S L, Tang L M, Zhao Y Q 2016 Acta Phys. Sin. 65 077301
Google Scholar
[17] 李立明, 宁锋, 唐黎明 2015 物理学报 64 227303
Google Scholar
Li L M, Ning F, Tang L M 2015 Acta Phys. Sin. 64 227303
Google Scholar
[18] Bernardi M, Palummo M, Grossman J C 2013 Nano Lett. 13 3664
Google Scholar
[19] Lopezsanchez O, Lembke D, Kayci M, Radenovic A, Kis A 2013 Nat. Nanotechnol. 8 497
Google Scholar
[20] Bertolazzi S, Brivio J, Kis A 2011 ACS Nano 5 9703
Google Scholar
[21] Eda G, Yamaguchi H, Voiry D, Fujita T, Chen M, Chhowalla M 2011 Nano Lett. 11 5111
Google Scholar
[22] Ning F, Wang D, Feng Y X, Tang L M, Zhang Y, Chen K Q 2017 J. Mater. Chem. C 5 9429
Google Scholar
[23] Cao T, Wang G, Han W, Ye H, Zhu C, Shi J, Niu Q, Tan P, Wang E, Liu B, Feng J 2012 Nat. Commun. 3 887
Google Scholar
[24] Li Q, Tang L, Zhang C, Wang D, Chen Q J, Feng Y X, Tang L M, Chen K Q 2017 Appl. Phys. Lett. 111 171602
Google Scholar
[25] Xu Y, Li Y, Chen X, Zhang C, Zhang R, Lu P 2016 AIP Adv. 6 075001
Google Scholar
[26] Kam K K, Parkinson B A 1982 J. Phys. Chem. 86 463
Google Scholar
[27] Chen J, Li S L, Xu Q, Tanaka K 2002 Chem. Commun. 16 1722
Google Scholar
[28] Cheng F Y, Chen J, Gou X L 2006 Adv. Mater. 18 2561
Google Scholar
[29] Karunadasa H I, Montalvo E, Sun Y, Majda M, Long J R, Chang C J 2012 Science 335 698
Google Scholar
[30] Mouri S, Miyauchi Y, Matsuda K 2013 Nano Lett. 13 5944
Google Scholar
[31] Chhowalla M, Amaratunga G A 2000 Nature 407 164
Google Scholar
[32] Muratore C, Voevodin A A 2006 Surf. Coat. Technol. 201 4125
Google Scholar
[33] Stefanov M, Enyashin A N, Heine T, Seifert G 2008 J. Phys. Chem. C 112 17764
Google Scholar
[34] Mak K F, Lee C, Hone J, Shan J, Heinz T F 2010 Phys. Rev. Lett. 105 136805
Google Scholar
[35] Elias D C, Nair R R, Mohiuddin T M G, Morozov S V, Blake P, Halsall M P, Ferrari A C, Boukhvalov D W, Katsnelson M I, Geim A K, Novoselov K S 2009 Science 323 610
Google Scholar
[36] Zou J, Tang L M, Chen K Q, Feng Y X 2017 J. Phys.: Condens. Matter 30 065001
Google Scholar
[37] Zhang W, Zhang Z, Yang W 2015 J. Nanosci. Nanotechnol. 15 8075
Google Scholar
[38] van der Marel D, Molegraaf H J A, Zaanen J, Nussinov Z, Carbone F, Damascelli H, Eisaki H, Greven M, Kes P H, Li M 2003 Nature 425 271
Google Scholar
[39] Sundberg P, Moyes R B, Tomkinson J 1991 Bull. Soc. Chim. Belg. 100 967
Google Scholar
[40] Lozada-Hidalgo M, Zhang S, Hu S, Kravets V G, Rodriguez F J, Berdyugin A, Grigorenko A, Geim A K 2018 Nat. Nanotechnol. 13 300
Google Scholar
[41] Perdew J P, Burke K, Ernzerhof M 1996 Phys. Rev. Lett. 77 3865
Google Scholar
[42] Blöchl P E 1994 Phys. Rev. B 50 17953
Google Scholar
[43] Henkelman G, Uberuaga B P, Jónsson H 2000 J. Chem. Phys. 113 9901
Google Scholar
[44] Koh E W K, Chiu C H, LimY K, ZhangY W, Pan H 2012 Int. J. Hydrogen Energy 37 14323
Google Scholar
[45] Özçelik V O, Azadani J G, Yang C, Koester S J, Low T 2016 Phys. Rev. B 94 035125
Google Scholar

 下载:
下载:
计量
- 文章访问数: 17595
- PDF下载量: 0
- 被引次数: 0
