











-
Based on the method of density functional theory B3LYP with a 6-311++G(d, p) basis set, the potential energy surface of conformational isomerization along the two-dimensional coordinates formed by the dihedral angles ϕ1(C9N1C2C5) and ϕ2(C16N1C9C12) in a range of –180°–180° is investigated. And 12 ground state conformers of triethylamine are identified. Furthermore,with the second-order Moller-Plesset perturbation theory MP2 on the same basis set level, the structures of six lower-energy conformers are optimized and their energy values are estimated. The results show that G1 and G1' with C3 symmetry are the most stable conformers and G4 and G4' with new methyl orientations are identified. In addition, some vibrational modes in the infrared spectra of G1–G4 are assigned and discussed. The infrared spectra of G1–G4 show that the intensity is weak in a range of 0–1600 cm–1, while the intensity is strong in a range of 2800–3300 cm–1. The characteristic vibration modes such as umbrella vibration and CH stretching vibration are assigned. The average shift of the corresponding infrared peaks on different conformations is estimated at less than 20 cm–1. -
Keywords:
- conformation isomerization /
- density-functional theory calculation /
- potential energy surface /
- infrared spectra
[1] Park S T, Kim S K, Kim M S 2002 Nature 415 306
 Google Scholar
Google Scholar
[2] Kim M H, Shen L, Tao H, Martinez T J, Suits A G 2007 Science 315 1561
Google Scholar
[3] Gosselin J L, Weber P M 2005 J. Phys. Chem. A 109 4899
Google Scholar
[4] Deb S, Bayes B A, Minitti M P, Weber P M 2011 J. Phys. Chem. A 115 1804
Google Scholar
[5] Minitti M P, Weber P M 2007 Phys. Rev. Lett. 98 253004
Google Scholar
[6] Kuthirummal N, Weber P M 2003 Chem. Phys. Lett. 378 647
Google Scholar
[7] Kuthirummal N, Weber P M 2006 J. Mol. Struct. 787 163
Google Scholar
[8] Dian B C, Clarkson J R, Zwier T S 2004 Science 303 1169
Google Scholar
[9] Kumar K 1971 Chem. Phys. Lett. 9 504
Google Scholar
[10] Crocker C, Goggin P L 1978 J. Chem. Soc. Dalton Trans 388
[11] Bushweller C H, Fleischman S H, Grady G L, McGoff P, Rithner C D, Whalon M R, Brennan J G, Marcantonio R P, Domingue R P 1982 J. Am. Chem. Soc. 104 6224
Google Scholar
[12] Fleischman S H, Weltin E E, Bushweller C H 1985 J. Comput. Chem. 6 249
Google Scholar
[13] Takeuchi H, Kojima T, Egawa T, Konaka S 1992 J. Phys. Chem. 96 4389
Google Scholar
[14] Grimme S 2011 Wiley Interdiscip. Rev. -Comput. Mol. Sci. 1 211
Google Scholar
[15] Grimme S, Hansen A, Brandenburg J G, Bannwarth C 2016 Chem. Rev. 116 5105
Google Scholar
[16] Sølling T I, Kötting C, Zewail A H 2003 J. Phys. Chem. A 107 10872
Google Scholar
[17] Cardoza J D, Rudakov F M, Weber P M 2008 J. Phys. Chem. A 112 10736
Google Scholar
[18] Deb S, Cheng X, Weber P M 2013 J. Phys. Chem. Lett. 4 2780
Google Scholar
[19] Cheng X, Zhang Y, Deb S, Minitti M P, Gao Y, Jónsson H, Weber P M 2014 Chem. Sci. 5 4394
Google Scholar
[20] Frisch M J, Trucks G W, Schlegel H B, Scuseria G E, Robb M A, Cheeseman J R, Scalmani G, Barone V, Mennucci B, Petersson G A, Nakatsuji H, Caricato M, Li X, Hratchian H P, Izmaylov A F, Bloino J, Zheng G, Sonnenberg J L, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Vreven T, Montgomery J A Jr, Peralta J E, Ogliaro F, Bearpark M, Heyd J J, Brothers E, Kudin K N, Staroverov V N, Kobayashi R, Normand J, Raghavachari K, Rendell A, Burant J C, Iyengar S S, Tomasi J, Cossi M, Rega N, Millam J M, Klene M, Knox J E, Cross J B, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann R E, Yazyev O, Austin A J, Cammi R, Pomelli C, Ochterski J W, Martin R L, Morokuma K, Zakrzewski V G, Voth G A, Salvador P, Dannenberg J J, Dapprich S, Daniels A D, Farkas Ö, Foresman J B, Ortiz J V, Cioslowski J, Fox D J 2009 Gaussian09 (Revision E.01)
[21] Vosko S H, Wilk L, Nusair M 1980 Can. J. Phys. 58 1200
Google Scholar
[22] Krishnan R, Binkley J S, Seeger R, Pople J A 1980 J. Chem. Phys. 72 650
Google Scholar
[23] Møller C, Plesset M S 1934 Phys. Rev. 46 618
Google Scholar
-

图 1 三乙胺分子在笛卡尔坐标系下的结构示意图.
Figure 1. Schematic structure of triethylamine in Cartesian coordinate system.

图 2 沿二面角ϕ1(C9N1C2C5)与ϕ2(C16N1C9C12)构成的二维坐标扫描–180°—180°范围内三乙胺构象异构化势能面
Figure 2. The conformational isomerization potential energy surface in the range of –180°–180° scanning along the two-dimensional coordinates formed by the dihedral angles of ϕ1 (C9N1C2C5) and ϕ2 (C16N1C9C12).

图 3 基于B3LYP/6-311++G(d, p)水平计算得到的三乙胺的12种稳定构象异构体的分子结构
Figure 3. The 12 stable conformers of triethylamine calculated on B3LYP/6-311++G(d, p) level.

图 4 ϕ1 (–100°—180°)与ϕ2 (40°—180°)范围内的6种构象异构体的势能面
Figure 4. The conformational isomerization potential energy surface of the six conformers in ϕ1(–100°–180°) and ϕ2 (40°–180°).
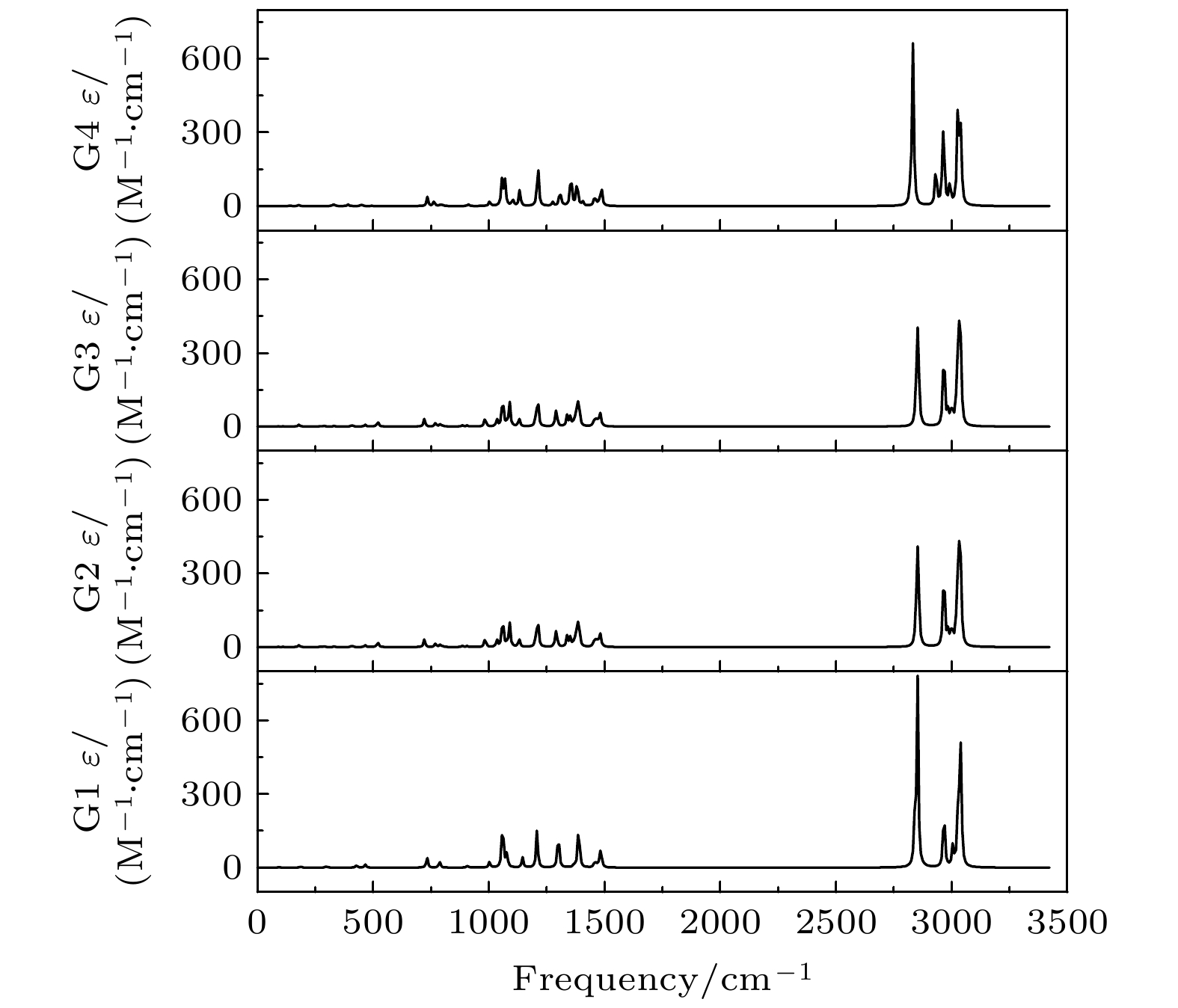
图 5 在B3LYP/6-311++G(d, p)水平上计算得到的G1—G4构象异构体的红外光谱
Figure 5. The infrared spectra of the G1–G4 conformers calculated at B3LYP/6-311++G(d, p) level.

图 6 基于B3LYP/6-311++G(d, p)水平计算得到的G1—G4构象异构体的伞形振动(上排图)和C—H对称伸缩振动(下排图)及其频率
Figure 6. The umbrella vibration mode (upper panel) and C—H symmetric stretch mode (lower panel) and their frequencies of the G1—G4 conformers calculated on B3LYP/6-311++G(d, p) level.
表 1 基于B3LYP/6-311++G(d, p)水平计算得到的三乙胺的12种稳定构象异构体的二面角与能量
Table 1. The energies and dihedral angles of the 12 conformers of triethylamine calculated on B3LYP/6-311++G(d, p) level
Conformers ϕ1/(°) ϕ2/(°) ϕ3/(°) Energy/Hartree Relative E/meV G1 (GGG) 155.42 155.60 155.64 –292.501669 0 G1' (G'G'G') 77.67 77.63 77.63 –292.501620 1.33 G2 (TG'G') –66.62 76.02 63.21 –292.500061 43.76 G3 (TGG) –64.76 165.17 151.59 –292.500061 43.76 G4 (GG'G'') 162.85 67.24 115.28 –292.499824 50.21 G4' (G'G''G) 67.24 115.26 162.86 –292.499824 50.21 G5 (G'GT) –167.71 –60.28 65.25 –292.499826 50.15 G6 (GGT) –75.99 –63.21 66.63 –292.500061 43.76 G7 (G'G'T) –165.17 –151.59 64.75 –292.500061 43.76 G8 (G'TG) 60.28 –65.24 167.73 –292.499826 50.15 G9 (GTG) 151.58 –64.74 165.19 –292.500061 43.76 G10 (TG'T) 56.64 –156.48 76.67 –292.497311 118.59  DownLoad: CSV
DownLoad: CSV
表 2 三乙胺的6种稳定构象异构体在MP2/6-311++G(d, p)计算下的二面角与能量
Table 2. The energies and dihedral angles of the six stable conformers of triethylamine on the level of MP2/6-311++G(d, p)
Conformers ϕ1/(°) ϕ2/(°) ϕ3/(°) Energy/Hartree Relative E/meV G1 (GGG) 157.62 157.63 157.63 –291.559737 0 G1' (G'G'G') 79.53 79.53 79.53 –291.559737 0 G2 (TG'G') –66.91 75.27 57.80 –291.558559 32.06 G3 (TGG) –59.62 175.87 158.42 –291.558563 31.95 G4 (GG'G'') 166.11 67.66 117.32 –291.558516 33.23 G4' (G'G''G) 67.74 117.11 166.19 –291.558517 33.20
DownLoad: CSV
表 3 基于B3LYP/6-311++G(d, p)水平计算得到的G1—G4构象异构体的振动模式与频率
Table 3. The vibrational modes and their frequencies of the G1-G4 conformers calculated on B3LYP/6-311++G(d, p) level
Modes Frequency /cm–1 Infrared /(arb. units) G1 G2 G3 G4 G1 G2 G3 G4 ν1 87.22 55.02 55.10 50.35 0.1532 0.0639 0.0639 0.0004 ν2 90.98 87.43 87.41 96.49 0.1978 0.1857 0.1857 0.0238 ν3 95.03 109.27 109.28 140.35 0.1084 0.2932 0.2933 0.7795 ν4 188.06 178.51 178.54 178.16 1.1011 2.0829 2.0830 1.1365 ν5 208.61 203.62 203.58 205.94 0.0793 0.0717 0.0724 0.0393 ν6 214.02 224.68 224.73 223.87 0.0468 0.0690 0.0687 0.1212 ν7 294.38 268.43 268.36 256.33 1.0986 0.3758 0.3757 0.1000 ν8 304.06 290.93 290.98 328.65 0.3330 1.0227 1.0225 1.6826 ν9 305.06 331.61 331.61 332.28 0.3301 0.6897 0.6908 0.3791 ν10 427.73 407.68 407.74 390.84 3.0184 1.9295 1.9286 1.8981 ν11 465.12 465.26 465.28 449.38 1.9245 2.3956 2.3948 1.6278 ν12 466.51 519.32 519.35 495.27 1.9272 6.0474 6.0464 0.2229 ν13 732.52 721.09 721.08 734.07 12.9402 9.4813 9.4797 10.9942 ν14 786.67 769.53 769.53 763.17 3.8307 4.3371 4.3412 6.1536 ν15 787.30 788.70 788.71 790.73 3.8921 2.6364 2.6346 1.2979 ν16 797.88 799.48 799.50 799.56 0.0602 1.6382 1.6374 1.5619 ν17 907.63 886.81 886.80 911.00 1.2513 1.6792 1.6806 1.3188 ν18 907.82 906.38 906.37 911.59 1.2450 1.3085 1.3102 0.5529 ν19 1002.59 983.42 983.41 1003.17 6.7677 11.0738 11.0739 5.7366 ν20 1059.24 1035.02 1035.00 1056.02 29.6466 8.2727 8.2697 18.7155 ν21 1059.73 1059.50 1059.46 1058.63 30.0288 30.8396 31.1034 16.0090 ν22 1065.72 1062.07 1062.06 1069.23 3.2100 8.4580 8.2166 30.8172 ν23 1078.10 1078.78 1078.77 1076.49 8.5384 3.6151 3.6028 1.3100 ν24 1078.68 1090.02 1090.04 1102.40 8.3190 28.7911 28.7831 8.1051 ν25 1145.20 1130.09 1130.09 1132.97 12.0682 10.1819 10.1824 19.8481 ν26 1208.09 1204.99 1204.96 1210.21 23.0883 18.4553 18.4412 17.6571 ν27 1208.58 1213.11 1213.11 1213.26 23.1735 25.1079 25.1196 35.4460 ν28 1295.98 1286.87 1286.86 1275.96 6.1543 5.5069 5.5346 4.4355 ν29 1299.90 1291.74 1291.73 1307.05 19.8870 19.0478 19.0085 20.9498 ν30 1301.08 1338.30 1338.29 1319.27 19.5949 13.3278 13.3106 1.2005 ν31 1368.21 1352.09 1352.09 1354.63 1.9917 11.3690 11.3924 37.8361 ν32 1369.16 1366.65 1366.64 1357.26 1.2772 3.0836 3.0821 5.3537 ν33 1377.64 1375.36 1375.35 1377.69 0.3191 3.9451 3.9459 0.5963 ν34 1388.04 1377.00 1377.00 1381.06 17.8509 14.4179 14.4269 19.6423 ν35 1388.15 1386.56 1386.55 1382.64 18.3740 20.6580 20.6282 12.8529 ν36 1388.87 1390.27 1390.26 1404.98 19.7944 15.6998 15.6974 5.4524 ν37 1457.17 1452.04 1452.05 1453.70 2.7473 4.8488 4.8565 5.2712 ν38 1457.42 1458.81 1458.81 1455.34 1.6935 4.5549 4.5301 0.3420 ν39 1457.69 1461.08 1461.08 1458.89 2.2572 2.7707 2.7867 3.6730 ν40 1462.71 1465.68 1465.67 1462.41 1.8527 2.0538 2.0584 4.0415 ν41 1470.31 1469.26 1469.25 1465.41 1.2048 3.4122 3.4014 0.0857 ν42 1470.92 1470.19 1470.18 1473.12 1.5336 1.3596 1.3783 3.1465 ν43 1480.41 1477.89 1477.88 1478.47 7.1590 2.9233 2.9249 1.5156 ν44 1481.40 1480.62 1480.60 1484.66 7.0152 14.8916 14.8851 10.1121 ν45 1485.83 1490.87 1490.86 1487.68 12.4306 1.4478 1.4478 14.3785 ν46 2840.18 2846.52 2846.55 2823.87 23.2447 36.5331 36.7500 30.6694 ν47 2840.74 2855.55 2855.62 2833.67 23.3082 133.4917 133.2390 190.3336 ν48 2852.73 2964.14 2964.10 2931.98 238.2268 28.9250 28.9420 52.7107 ν49 2966.71 2965.86 2965.83 2961.43 29.7433 22.5935 22.5842 24.4676 ν50 2967.27 2967.80 2967.77 2962.15 29.7303 26.9332 26.9495 32.0017 ν51 2967.73 2969.71 2969.69 2966.69 20.4277 26.6680 26.6499 55.0959 ν52 3005.55 2985.78 2985.82 2967.51 12.9188 19.0174 18.9607 9.9999 ν53 3006.00 2999.96 2999.91 2982.63 12.0858 16.2484 16.2880 1.3433 ν54 3009.33 3003.92 3003.90 2993.29 0.0669 7.0967 7.1223 29.3355 ν55 3025.88 3022.39 3022.35 3025.08 9.0331 39.4849 39.4879 51.7362 ν56 3028.87 3026.72 3026.71 3026.92 48.6932 15.0249 15.0950 24.4990 ν57 3029.12 3029.64 3029.64 3028.60 44.4949 61.7552 61.8929 54.1919 ν58 3037.74 3031.73 3031.72 3036.45 53.6899 42.2463 42.0251 3.3194 ν59 3039.70 3037.26 3037.24 3037.37 45.2729 52.3351 52.2987 69.7071 ν60 3040.26 3040.20 3040.20 3041.40 46.0316 44.4987 44.5093 31.7214
DownLoad: CSV
-
[1] Park S T, Kim S K, Kim M S 2002 Nature 415 306
Google Scholar
[2] Kim M H, Shen L, Tao H, Martinez T J, Suits A G 2007 Science 315 1561
Google Scholar
[3] Gosselin J L, Weber P M 2005 J. Phys. Chem. A 109 4899
Google Scholar
[4] Deb S, Bayes B A, Minitti M P, Weber P M 2011 J. Phys. Chem. A 115 1804
Google Scholar
[5] Minitti M P, Weber P M 2007 Phys. Rev. Lett. 98 253004
Google Scholar
[6] Kuthirummal N, Weber P M 2003 Chem. Phys. Lett. 378 647
Google Scholar
[7] Kuthirummal N, Weber P M 2006 J. Mol. Struct. 787 163
Google Scholar
[8] Dian B C, Clarkson J R, Zwier T S 2004 Science 303 1169
Google Scholar
[9] Kumar K 1971 Chem. Phys. Lett. 9 504
Google Scholar
[10] Crocker C, Goggin P L 1978 J. Chem. Soc. Dalton Trans 388
[11] Bushweller C H, Fleischman S H, Grady G L, McGoff P, Rithner C D, Whalon M R, Brennan J G, Marcantonio R P, Domingue R P 1982 J. Am. Chem. Soc. 104 6224
Google Scholar
[12] Fleischman S H, Weltin E E, Bushweller C H 1985 J. Comput. Chem. 6 249
Google Scholar
[13] Takeuchi H, Kojima T, Egawa T, Konaka S 1992 J. Phys. Chem. 96 4389
Google Scholar
[14] Grimme S 2011 Wiley Interdiscip. Rev. -Comput. Mol. Sci. 1 211
Google Scholar
[15] Grimme S, Hansen A, Brandenburg J G, Bannwarth C 2016 Chem. Rev. 116 5105
Google Scholar
[16] Sølling T I, Kötting C, Zewail A H 2003 J. Phys. Chem. A 107 10872
Google Scholar
[17] Cardoza J D, Rudakov F M, Weber P M 2008 J. Phys. Chem. A 112 10736
Google Scholar
[18] Deb S, Cheng X, Weber P M 2013 J. Phys. Chem. Lett. 4 2780
Google Scholar
[19] Cheng X, Zhang Y, Deb S, Minitti M P, Gao Y, Jónsson H, Weber P M 2014 Chem. Sci. 5 4394
Google Scholar
[20] Frisch M J, Trucks G W, Schlegel H B, Scuseria G E, Robb M A, Cheeseman J R, Scalmani G, Barone V, Mennucci B, Petersson G A, Nakatsuji H, Caricato M, Li X, Hratchian H P, Izmaylov A F, Bloino J, Zheng G, Sonnenberg J L, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Vreven T, Montgomery J A Jr, Peralta J E, Ogliaro F, Bearpark M, Heyd J J, Brothers E, Kudin K N, Staroverov V N, Kobayashi R, Normand J, Raghavachari K, Rendell A, Burant J C, Iyengar S S, Tomasi J, Cossi M, Rega N, Millam J M, Klene M, Knox J E, Cross J B, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann R E, Yazyev O, Austin A J, Cammi R, Pomelli C, Ochterski J W, Martin R L, Morokuma K, Zakrzewski V G, Voth G A, Salvador P, Dannenberg J J, Dapprich S, Daniels A D, Farkas Ö, Foresman J B, Ortiz J V, Cioslowski J, Fox D J 2009 Gaussian09 (Revision E.01)
[21] Vosko S H, Wilk L, Nusair M 1980 Can. J. Phys. 58 1200
Google Scholar
[22] Krishnan R, Binkley J S, Seeger R, Pople J A 1980 J. Chem. Phys. 72 650
Google Scholar
[23] Møller C, Plesset M S 1934 Phys. Rev. 46 618
Google Scholar
-
[1] Tong Zan, Yang Yin-Li, Xu Jing, Liu Wei, Chen Liang. Theoretical study of helium separation performance of crown ether-graphane membranes. Acta Physica Sinica, 2023, 72(6): 068201. doi: 10.7498/aps.72.20222183 [2] Cui Zi-Chun, Yang Mo-Han, Ruan Xiao-Peng, Fan Xiao-Li, Zhou Feng, Liu Wei-Min. High-throughput calculation of interfacial friction of two-dimensional material. Acta Physica Sinica, 2023, 72(2): 026801. doi: 10.7498/aps.72.20221676 [3] Qiu Zi-Yang, Chen Yan, Qiu Xiang-Gang. Infrared spectroscopic study of topological material BaMnSb2. Acta Physica Sinica, 2022, 71(10): 107201. doi: 10.7498/aps.71.20220011 [4] Li Wen-Tao, Yuan Mei-Ling, Wang Jie-Min. Dynamics of C+ + H2 reaction based on a new potential energy surface. Acta Physica Sinica, 2022, 71(9): 093402. doi: 10.7498/aps.71.20212241 [5] Shi Bin, Yuan Li, Tang Tian-Yu, Lu Li-Min, Zhao Xian-Hao, Wei Xiao-Nan, Tang Yan-Lin. Spectral analysis and density functional theory study of tert-butylhydroquinone. Acta Physica Sinica, 2021, 70(5): 053102. doi: 10.7498/aps.70.20201555 [6] Zhao Wen-Li, Wang Yong-Gang, Zhang Lu-Lu, Yue Da-Guang, Meng Qing-Tian. Wave packet quantum dynamics of ${\bf{C}}{(^3}{\bf{P}}) + {{\bf{H}}_2}({{\bf{X}}^1} \Sigma _{\bf{g}}^ + ) $ $ \to {\bf{H}}{(^2}{\bf{S}}) + {\bf{CH}}{(^2} \Pi ) $ reaction based on new CH2(${\tilde {\bf X}{}^3}\bf A''$ ) surface. Acta Physica Sinica, 2020, 69(8): 083401. doi: 10.7498/aps.69.20200132[7] Wu Chen-Chen, Guo Xiang-Dong, Hu Hai, Yang Xiao-Xia, Dai Qing. Graphene plasmon enhanced infrared spectroscopy. Acta Physica Sinica, 2019, 68(14): 148103. doi: 10.7498/aps.68.20190903 [8] Xu Bing, Qiu Zi-Yang, Yang Run, Dai Yao-Min, Qiu Xiang-Gang. Optical properties of topological semimetals. Acta Physica Sinica, 2019, 68(22): 227804. doi: 10.7498/aps.68.20191510 [9] Wang An-Jing, Fang Yong-Hua, Li Da-Cheng, Cui Fang-Xiao, Wu Jun, Liu Jia-Xiang, Li Yang-Yu, Zhao Yan-Dong. Simulation of pollutant-gas-cloud infrared spectra under plane-array detecting. Acta Physica Sinica, 2017, 66(11): 114203. doi: 10.7498/aps.66.114203 [10] Chi Bao-Qian, Liu Yi, Xu Jing-Cheng, Qin Xu-Ming, Sun Chen, Bai Cheng-Hao, Liu Yi-Fan, Zhao Xin-Luo, Li Xiao-Wu. Density functional theory study of structure stability and electronic structures of graphyne derivatives. Acta Physica Sinica, 2016, 65(13): 133101. doi: 10.7498/aps.65.133101 [11] Yang Xue, Yan Bing, Lian Ke-Yan, Ding Da-Jun. Theoretical study on the photodissociation reaction of α-cyclohexanedione in ground state. Acta Physica Sinica, 2015, 64(21): 213101. doi: 10.7498/aps.64.213101 [12] Han Yu-Long, Li Zhen, Wang Jiang-Hong, Feng Er-Yin, Huang Wu-Ying. Potential energy surface and spectra prediction for the Mg-CO complex. Acta Physica Sinica, 2013, 62(9): 093101. doi: 10.7498/aps.62.093101 [13] Li Xin, Yang Meng-Shi, Ye Zhi-Peng, Chen Liang, Xu Can, Chu Xiu-Xiang. DFT research on the IR spectrum of glycine tryptophan oligopeptides chain. Acta Physica Sinica, 2013, 62(15): 156103. doi: 10.7498/aps.62.156103 [14] Liu Xiao-Dong, Tao Wan-Jun, Hagihala Masato, Guo Qi-Xin, Meng Dong-Dong, Zhang Sen-Lin, Zheng Xu-Guang. Mid-infrared spectroscopic properties of geometrically frustrated basic cobalt chlorides. Acta Physica Sinica, 2011, 60(3): 037803. doi: 10.7498/aps.60.037803 [15] Lu Zhen-Ping, Han Kui, Li Hai-Peng, Zhang Wen-Tao, Huang Zhi-Min, Shen Xiao-Peng, Zhang Zhao-Hui, Bai Lei. Theoretical study of molecular vibrational hyperpolarizability of 4-N-methylstilbazonium salt derivatives. Acta Physica Sinica, 2007, 56(10): 5843-5848. doi: 10.7498/aps.56.5843 [16] Yu Chun-Ri, Feng Er-Yin, Cheng Xin-Lu, Yang Xiang-Dong. Theoretical study of the potential energy surface and differential scattering cross sections of He-HI complex. Acta Physica Sinica, 2007, 56(8): 4441-4447. doi: 10.7498/aps.56.4441 [17] Jiang Zhen-Yi, Li Sheng-Tao. First principles study of potential energy curves of NiTi alloy. Acta Physica Sinica, 2006, 55(11): 6032-6035. doi: 10.7498/aps.55.6032 [18] Han Hui-Xian, Peng Qian, Wen Zhen-Yi, Wang Yu-Bin. Local potential energy surface and vibration analysis for the S2O molecule. Acta Physica Sinica, 2005, 54(1): 78-84. doi: 10.7498/aps.54.78 [19] Yang Wu-Bao, Wang Jiu-Li, Zhang Gu-Ling, Fan Song-Hua, Liu Chi-Zi, Yang Si-Ze. Diamond-like carbon films deposited on optical glass substrate by using ECR microwave acetone plasma CVD method. Acta Physica Sinica, 2004, 53(9): 3099-3103. doi: 10.7498/aps.53.3099 [20] Wang Xiao-Yan, Ding Shi-Liang. Constructing potential energy surface of tetratomic molecules using Lie algebra. Acta Physica Sinica, 2004, 53(2): 423-426. doi: 10.7498/aps.53.423

 DownLoad:
DownLoad:



Catalog
Metrics
- Abstract views: 11328
- PDF Downloads: 196
- Cited By: 0
