










-
为了更好地在Si衬底上外延生长GaN薄膜, 需要先生长缓冲层(如AlN), 其中能否对起始的金属Al层实现可控生长, 将决定最终外延层的材料质量. 本文采用基于密度泛函理论的第一性原理计算, 理论上模拟计算了金属Al原子分别在清洁的、H原子和Cl原子钝化的Si(100)及Si(111)表面的吸附及扩散动力学行为. 研究结果显示, 在清洁的Si(100)表面上, Al原子易于吸附在沟槽中Tr位点, 沿沟槽呈曲折状扩散; 在H钝化、Cl钝化的Si(100)表面上, Al原子易于吸附在二聚体列顶部的H位置, 在二聚体列顶部沿直线扩散. 在不同方式处理的Si(111)表面, Al原子的最稳定吸附位置相同, 均易吸附于第二层Si原子的Top位(T4位点), 扩散路径类似, 均沿T4到H3 (空心位点)的路径扩散. 无论是Si(100)还是Si(111)表面, H钝化、Cl钝化处理Si表面均有效降低Al原子的扩散能垒, 使Al原子更容易在二维表面上扩散, 并通过吸附能的比较以及差分电荷密度图分析, 讨论了不同Si表面状态对金属Al原子吸附和扩散行为调制的物理机制.
Density functional theory is used to calculate the adsorption and diffusion behavior of Al atoms on clean, H-terminate, Cl-terminate Si(100) and Si(111) surfaces. The most stable position of Al atom adsorption and the diffusion path are different on Si(100) surface terminated by different methods. On the surface of clean Si(100), the Tr site is the most stable site for Al atom with an adsorption energy of 4.01 eV, and the H and M sites are the sub-stable stable sites with the adsorption energies of 3.51 eV, and 3.63 eV, respectively. When the Al atom is adsorbed at the Tr site on the clean Si(100) surface, it bonds with the Si atom to destroy the Si—Si bond in the dimer. Therefore Al is easily adsorbed at the Tr site of the trench and diffuses in a zigzag pattern along the trench. On the H-terminate and Cl-terminate Si(100) surface, Si—Si bonds in the dimer column are changed from cross to parallel. Al is easily adsorbed at the H position at the top of the dimer column, and diffuses along the line at the top of the dimer. The differential charge density shows that the Al atom transfers electrons to the Si atoms on the surface, and the surface H-terminate and Cl-terminate weaken the interaction between Al atoms and Si, and reduces the diffusion energy barrier of Al atoms. The Si(111) surface terminated by different methods has the same stable position (T4 site) for the adsorption of Al atoms. When Al atom adsorbs at the T4 site on the clean Si(100) surface, it bonds to Si atom, which located at the three T1 site, then Al atom is firmly fixed by the three Al—Si bonds with a bond length of 2.55 Å. Thus Al atom can has the largest adsorption energy and form the most stable state at the T1 site. With the diffusion and migration of Al atom, the bond between Al atom and the T1 site in the opposite direction appears to be broken. When Al atom migrating to the saddle point position is the most unstable. Here Al atom bonds to the Si atoms of the two adjacent T1 sites to form a bond with a length of 2.49 Å, which is 0.06 Å shorter than the initial Al—Si bond (2.55 Å). What’s more, the diffusion energy barrier of Al atom at this position is 0.65 eV, which impede Al atom to diffuse and migrate. When Al atom migrates to the H site, it rebonds to the three Si atoms on the adjacent surface and forms a bond with a length of 2.52 Å, which is 0.03 Å shorter than the Al—Si bond (2.55 Å) at the initial position. On the H-terminate and Cl-terminate Si(111) surface, Al atom doesn’t bond with Si atom for the H or Cl saturates the dangling bonds on the Si surface. The Si(111) surface terminated by different methods has the same stable position for adsorption of Al atoms. The diffusion paths of Al atoms are similar, and they are easy to be adsorbed to the top position (T4 site) of the second Si atom, and the path along T4 to H3 is diffused. Similarly, the H-terminate or Cl-terminate of Si(111) surface weakens the electron transfer between Al and Si atoms and reduces the diffusion energy barrier of Al atoms. Regardless of the Si(100) or Si(111) surface, the H-terminate and Cl-terminate Si surfaces are effective in reducing the diffusion barrier of Al atoms. -
Keywords:
- Si surfaces /
- Al /
- density functional theory /
- adsorption /
- diffusion
[1] Bak S J, Mun D H, Jung K C, et al. 2013 Electron. Mater. Lett. 9 367
 Google Scholar
Google Scholar
[2] Semond F 2015 MRS Bull. 40 412
Google Scholar
[3] Liu X Y, Li H F, Uddin A, et al. 2007 J. Cryst. Growth 300 114
Google Scholar
[4] Cheng K, Leys M, Degroote S, et al. 2006 J. Electron. Mater. 35 592
Google Scholar
[5] Yuan Y, Zuo R, Mao K, et al. 2018 Appl. Surf. Sci. 436 50
Google Scholar
[6] Cordier Y, Comyn R, Frayssinet E, et al. 2018 Phys. Status Solidi A 215 1700637
Google Scholar
[7] Zhao D, Zhao D 2018 J. Semicond. 39 033006
Google Scholar
[8] Wang X, Li H, Wang J, et al. 2014 Electron. Mater. Lett. 10 1069
Google Scholar
[9] Liu B T, Ma P, Li X L, et al. 2017 Chin. Phys. Lett. 34 058101
Google Scholar
[10] Novák T, Kostelník P, Konečný M, et al. 2019 Jpn. J. Appl. Phys. 5 SC1018
[11] Lee S J, Jeon S R, Ju J W, et al. 2019 J. Nanosci. Nanotech. 19 892
Google Scholar
[12] Cao M S, Shu J C, Wang X X, et al. 2019 Ann. Phys. 531 1800390
Google Scholar
[13] Cao M S, Wang X X, Zhang M, et al. 2019 Adv. Funct. Mater. 29 1807398
Google Scholar
[14] Zhang M, Wang X X, Cao W Q, et al. 2019 Adv. Opt. Mater. 1900689
Google Scholar
[15] Albao M A, Hsu C H, Putungan D B, et al. 2010 Surf. Sci. 604 396
Google Scholar
[16] Luniakov Y V 2011 Surf. Sci. 605 1866
Google Scholar
[17] Matsuo Y, Kangawa Y, Togashi R, et al. 2007 J. Cryst. Growth 300 66
Google Scholar
[18] Kresse G 1993 Phys. Rev. B 47 558
Google Scholar
[19] Kresse G, Furthmüller J 1996 Phys. Rev. B 54 11169
Google Scholar
[20] Stadler R, Podloucky R, Kresse G, et al. 1998 Phys. Rev. B 57 4088
Google Scholar
[21] Henkelman G, Jónsson H 2000 J. Chem. Phys. 113 9978
Google Scholar
[22] Sheppard D, Terrell R, Henkelman G 2008 J. Chem. Phys. 128 134106
Google Scholar
[23] Sheppard D, Henkelman G 2011 J. Comput. Chem. 32 1769
Google Scholar
[24] Ly D Q, Paramonov L, Makatsoris C 2009 J. Phys.: Condens. Matter. 21 185006
Google Scholar
[25] Kepenekian M, Robles R, Rurali R, et al. 2014 Nanotechnology 25 465703
Google Scholar
[26] Hsieh M F, Lin D S, Tsay S F 2009 Phys. Rev. B 80 045304
Google Scholar
-
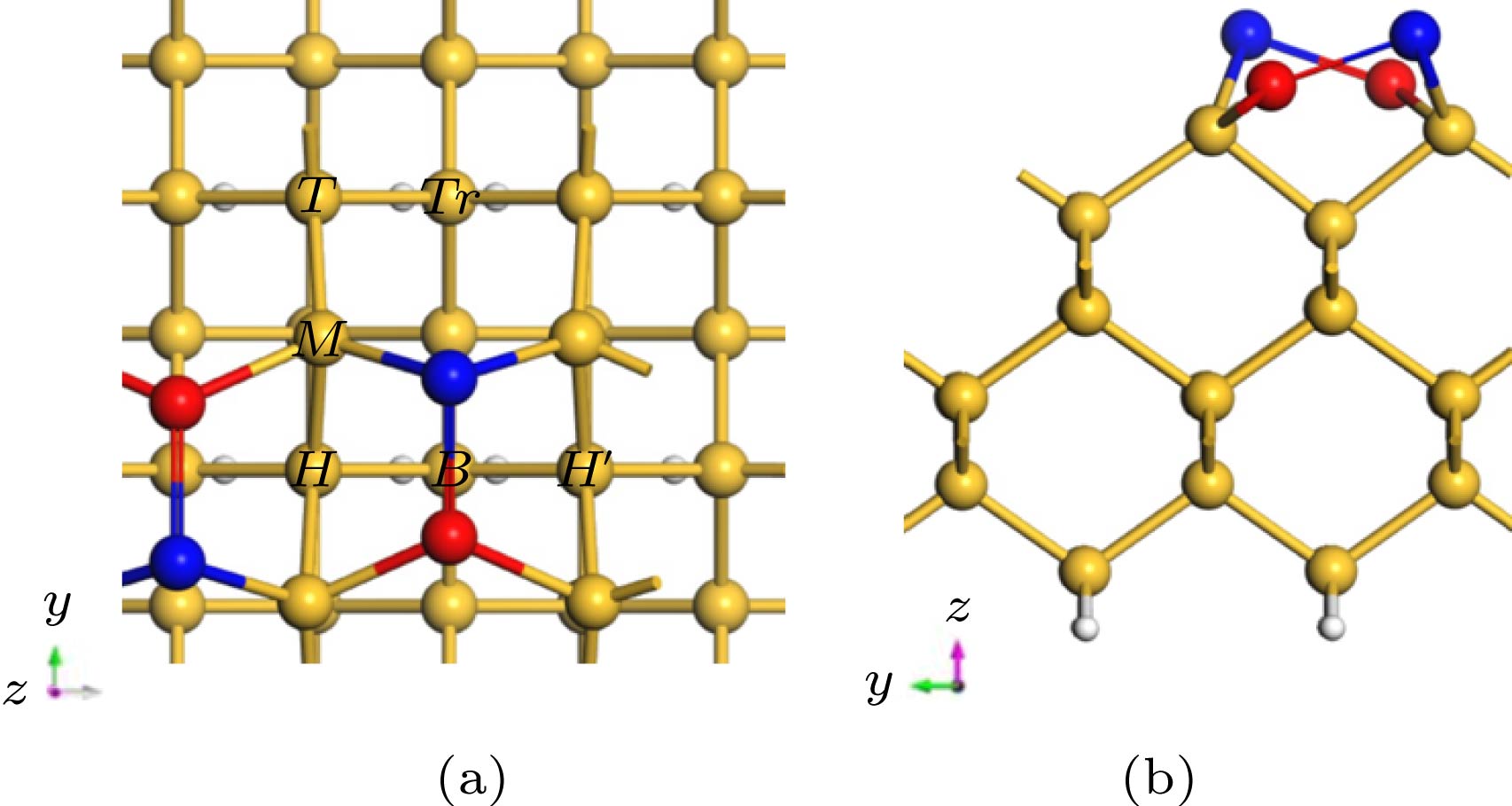
图 1 Si(100)表面的几何结构, 其中深蓝色和红色用于标记Si-Si二聚体中高和低两种位置的Si原子 (a)俯视图; (b)侧视图
Fig. 1. The geometry of the Si(100) surface, in which dark blue and red are used to mark the Si atoms in the high and low positions in the Si-Si dimer: (a) Top view; (b) side view.
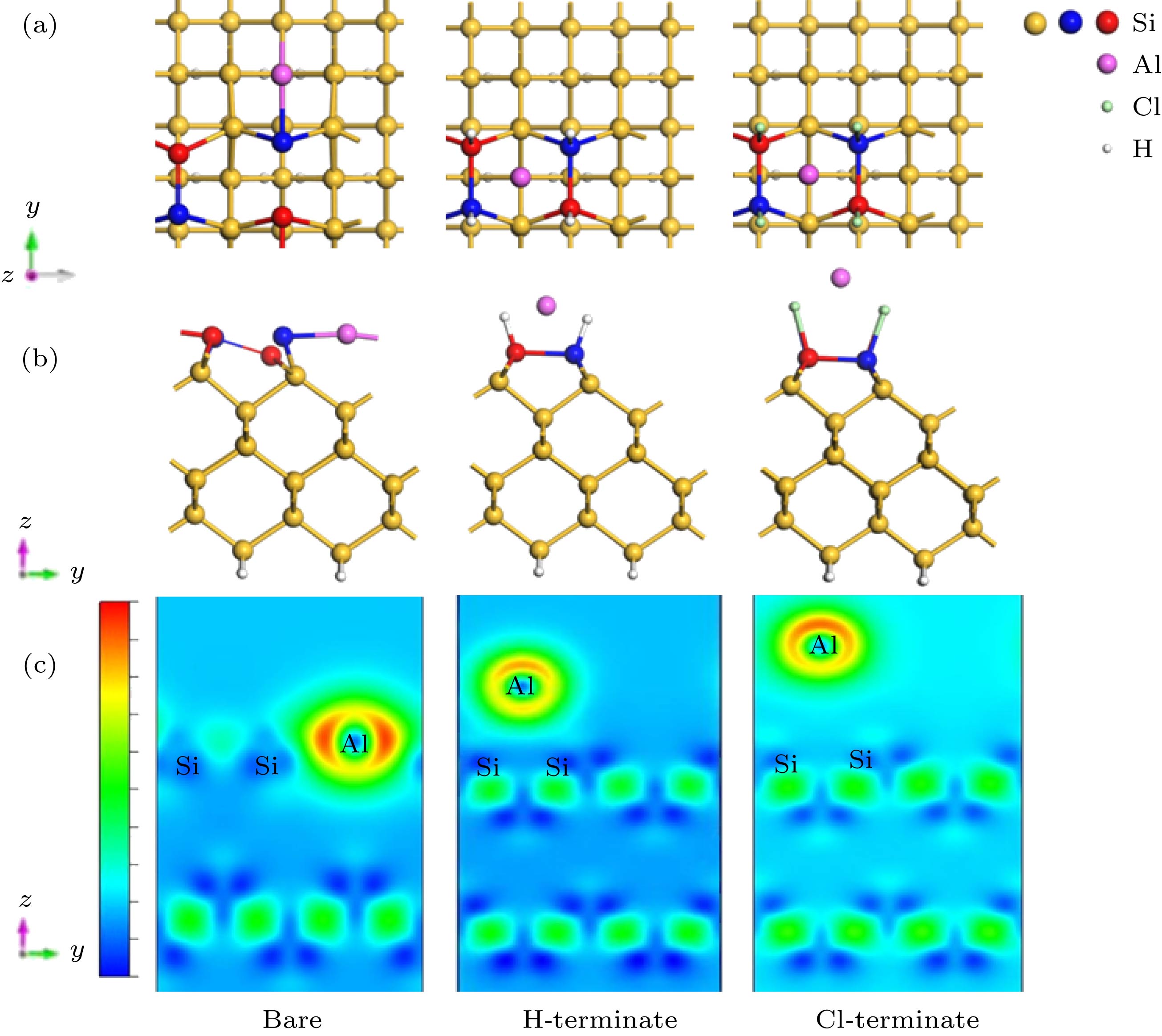
图 2 Al原子吸附在清洁Si(100) Tr位点、H钝化Si(100) H位点和Cl钝化Si(100) 表面H位点 (a)俯视图; (b)侧视图; (c)差分电荷密度图
Fig. 2. Al atom adsorption on the clean Si (100) Tr site, H-terminate Si (100) H site and Cl-terminate Si (100) surface H site: (a) Top view; (b) side view; (c) differential charge image.
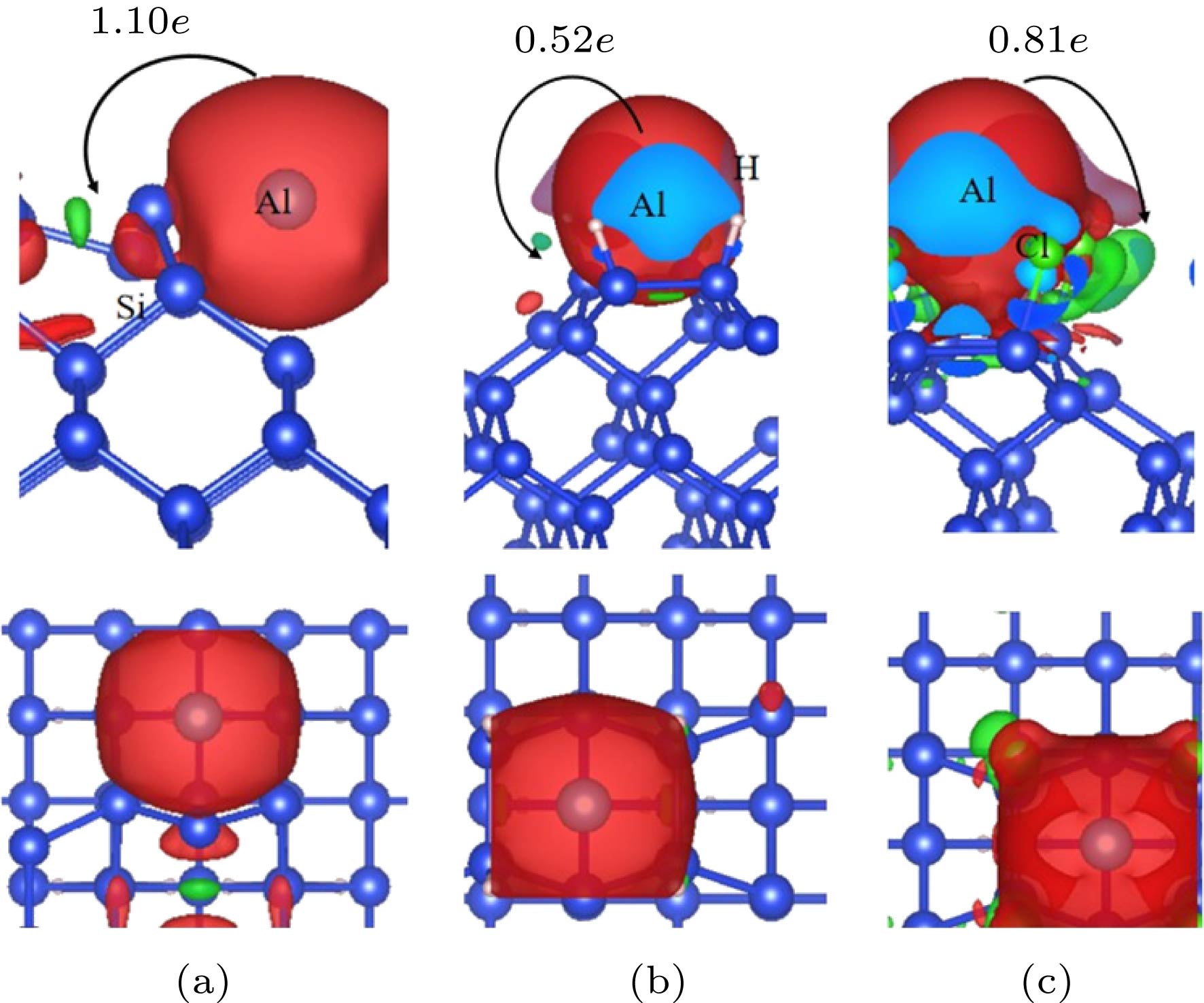
图 3 Al原子吸附在(a)清洁、(b)氢化、(c)氯化Si(100)表面后差分电荷图和Bader电子转移情况
Fig. 3. Bader charge transfer for Al atom adsorption on (a) the clean Si (100) Tr site, (b) H-terminate Si (100) H site and (c) Cl-terminate Si (100) surface H site.
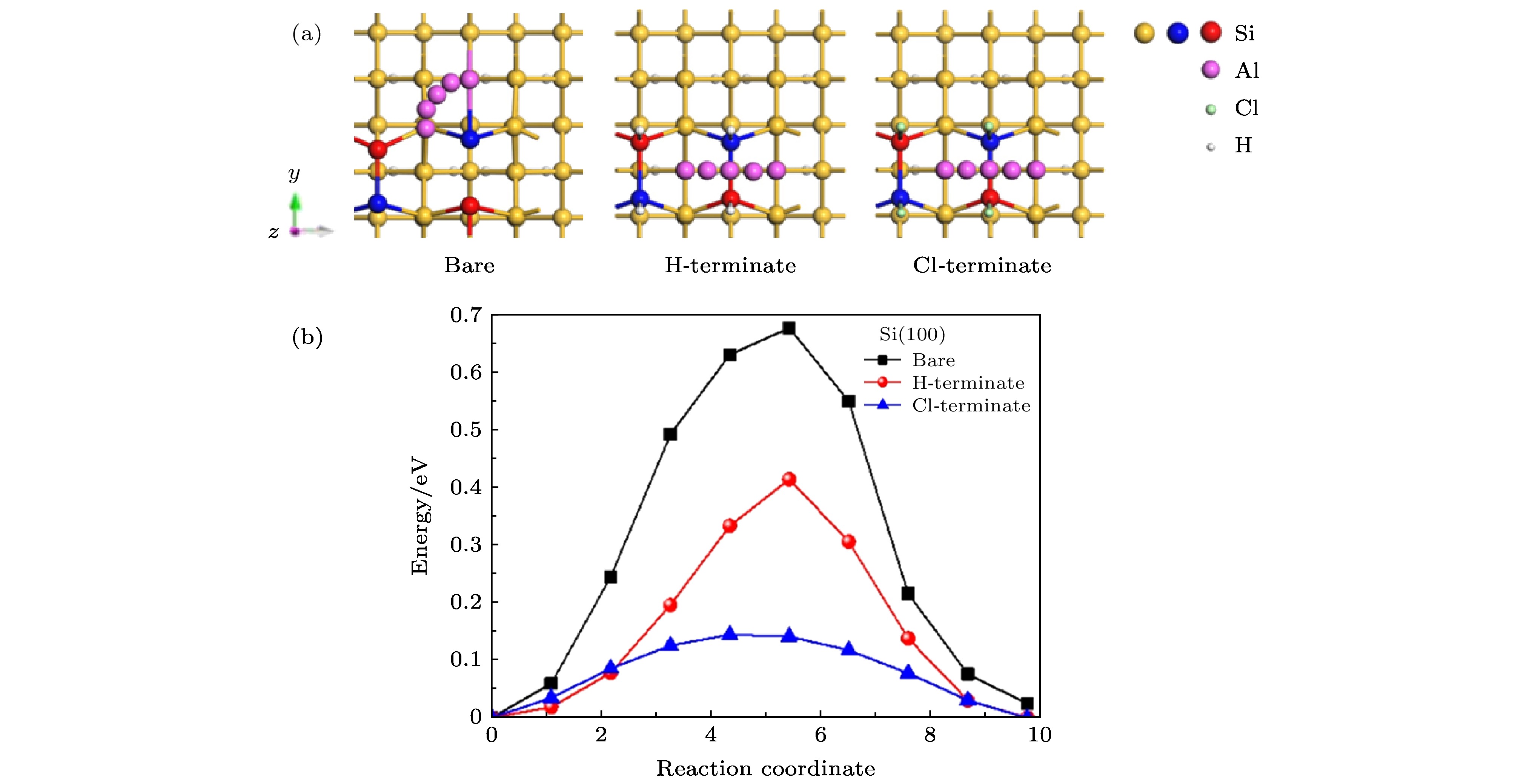
图 4 Al原子在清洁、H钝化、Cl钝化Si(100)表面 (a)扩散路径和; (b)扩散能垒
Fig. 4. Al atoms on clean, H-terminate and Cl-terminate Si(100) surfaces: (a) Diffusion paths; (b) diffusion barriers.
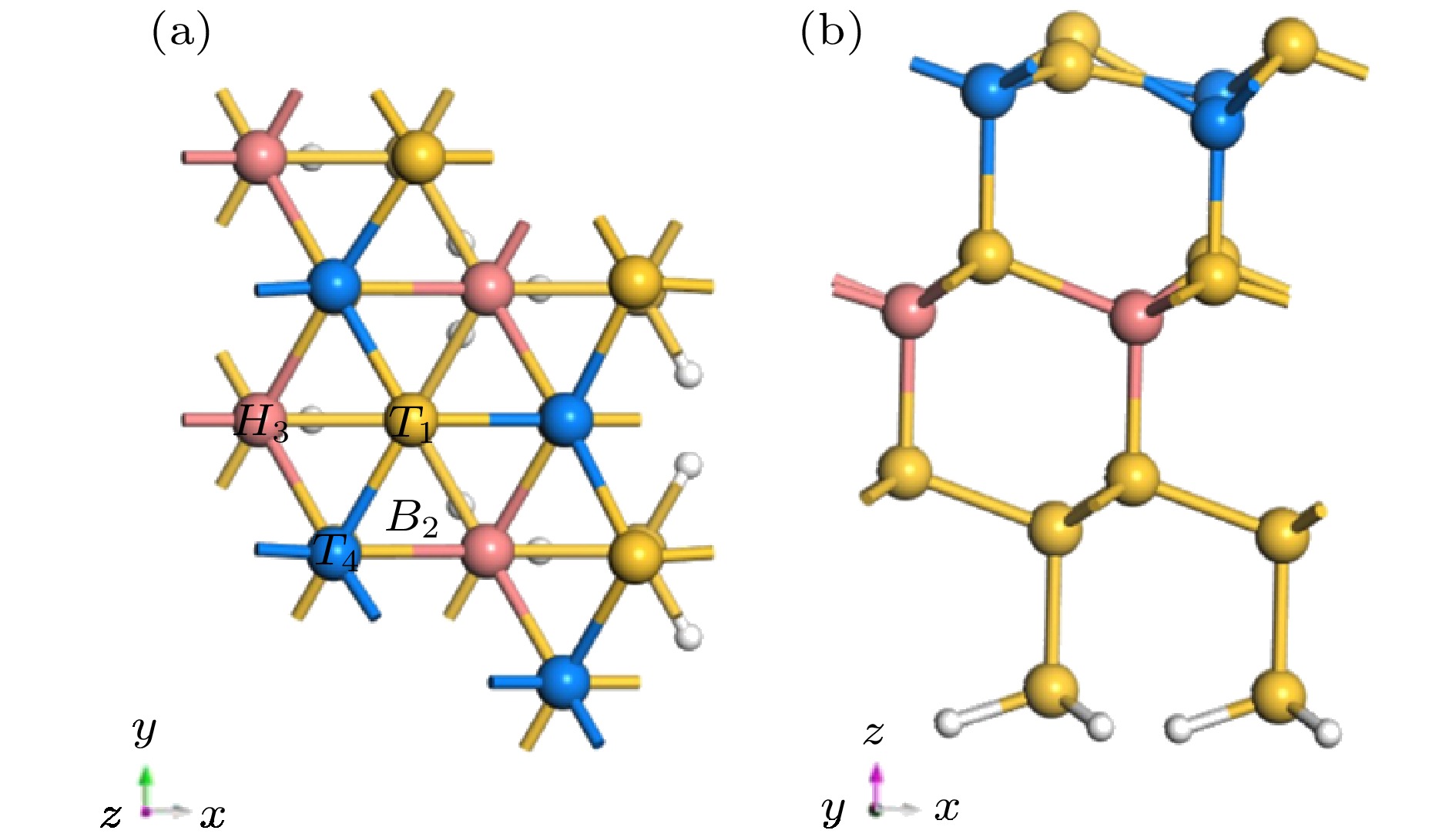
图 5 (a)和(b)分别为清洁Si(111)表面俯视图和侧视图T1, B2, H3和T4为Al原子在Si(111)表面的吸附的四个高对称位
Fig. 5. (a) and (b) are the top and side views of the clean Si (111) surface, respectively; T1, B2, H3 and T4 are the four highly symmetric sites of adsorption of Al atoms on the Si(111) surface.
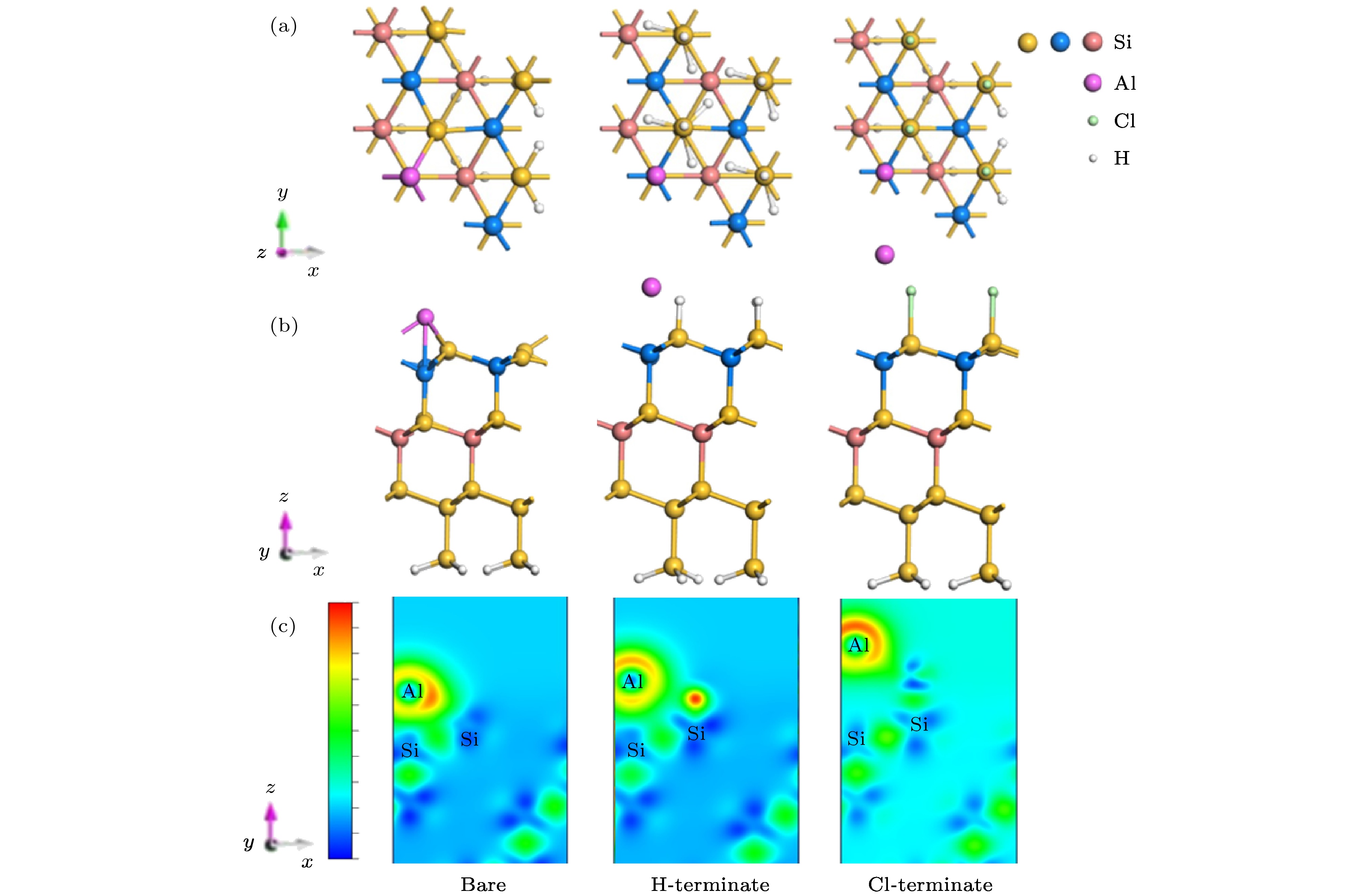
图 6 Al原子吸附在清洁、H钝化、Cl钝化Si(111)表面的(a)俯视图, (b)侧视图, (c)差分电荷密度图
Fig. 6. Al atom adsorbed on clean, H-terminate, Cl-terminate Si(111) surface: (a) Top view; (b) side view; (c) differential charge image
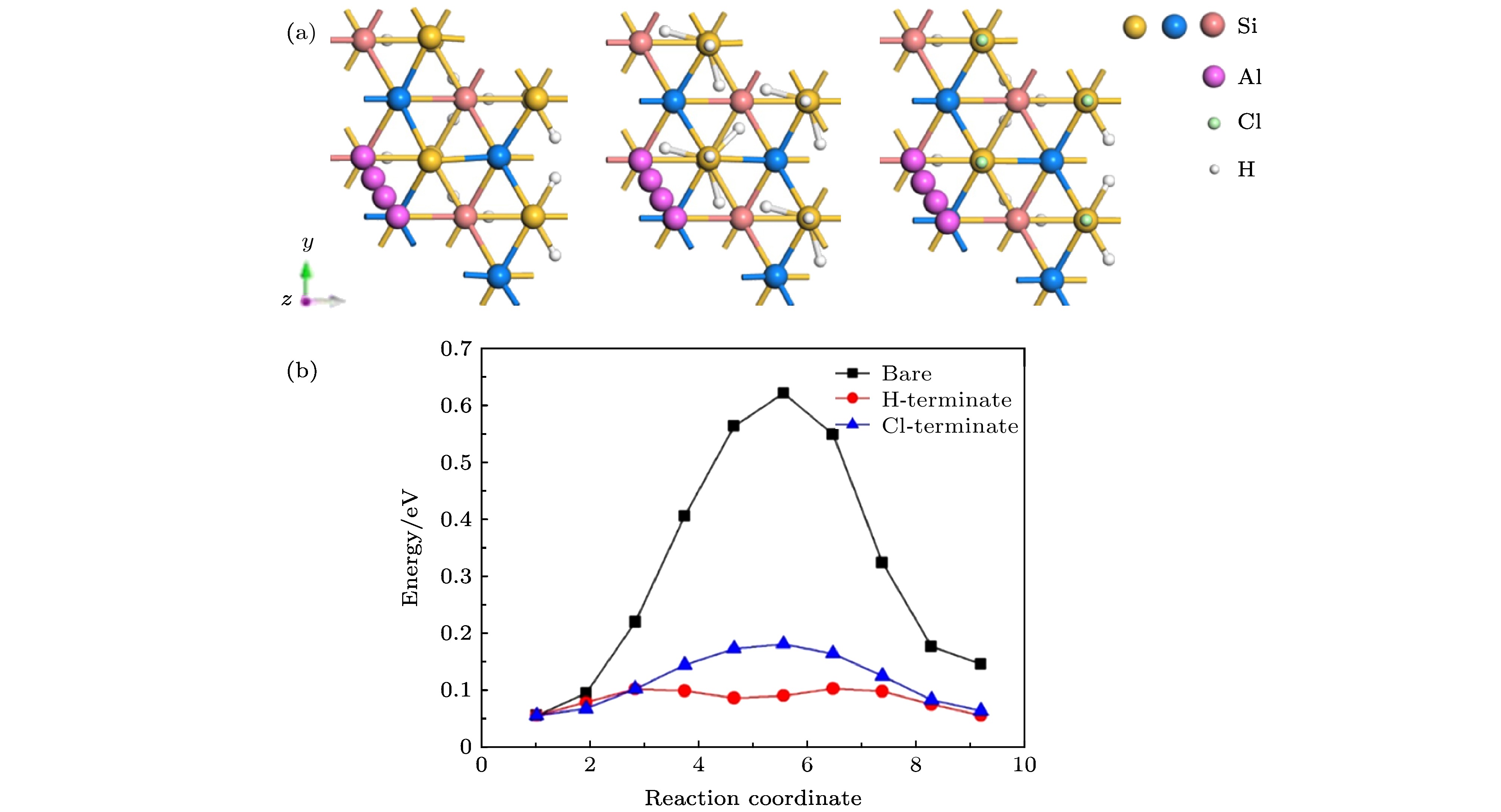
图 7 Al原子在清洁、H钝化、Cl钝化Si(111)表面 (a)扩散路径; (b)扩散能垒
Fig. 7. Al atoms on clean, H-terminate, and Cl-terminate Si(111) surfaces: (a) Diffusion paths; (b) diffusion barriers.
表 1 Al原子吸附在清洁、H钝化和Cl钝化Si(100)表面的吸附能Ead(eV)和结构参数; 其中Si—Si为二聚体Si—Si的键长, Si—Aladjacent为Al原子与邻近Si原子之间的距离
Table 1. Adsorption energy Ead (eV) and structural parameters of Al atom adsorbed on clean, H-terminate and Cl-terminate Si(100) surface; Si—Si is the bond length of dimer Si—Si, Si—Aladjacent is the distance between the Al atom and the adjacent Si atom.
Si(100) surface Site Eads/eV Si—Si/Å Si—Aladjacent/Å Bare Tr 4.01 2.77 2.45 M 3.96 2.43 2.52 H 3.63 2.45 2.45 H-terminate Tr 1.54 2.41 2.69 T 1.50 2.40 2.69 H 1.37 2.40 2.98 Cl-terminate T 2.03 2.42 2.87 M 1.17 2.40 1.17 B 2.04 2.42 4.09 H 1.80 2.39 2.83  下载: 导出CSV
下载: 导出CSV
表 3 Al原子吸附在清洁、H钝化、Cl钝化Si(100)和Si(111)表面的扩散路径和扩散能垒
Table 3. Diffusion path and diffusion energy of Al atom adsorbed on clean, H-terminate, Cl-terminate Si(100) and Si(111) surfaces.
Type Si(100) Si(111) Bare H-terminate Cl-terminate Bare H-terminate Cl-terminate Path Tr → M H → $H'$ H → $H'$ T4 → H3 T4 → H3 T4 → H3 Eads/eV 0.60 0.38 0.13 0.65 0.05 0.14
下载: 导出CSV
表 2 Al原子吸附在清洁、H钝化、Cl钝化Si(111)的几何位点, 其中Eads为对应位点的吸附能, d1, d2, d3为Al原子与邻近三个Si原子之间的距离, h为竖直方向Al原子与邻近Si原子之间的距离
Table 2. Al atom adsorption in the clean, H-terminate, Cl-terminate Si (111) geometric site, where Eads is the adsorption energy of the adjacent site; d1, d2 and d3 are the distance between the Al atom and the adjacent three Si atoms, h is the distance between the Al atom and the adjacent Si atom in the vertical direction.
Si(111) surface Site Eads/eV d1/Å d2/Å d3/Å h/Å Bare T4 4.51 2.55 2.55 2.55 2.57 H3 4.35 2.52 2.52 2.52 5.37 H-terminate T4 1.36 3.09 3.06 3.05 2.71 H3 1.13 2.93 2.95 2.92 5.76 Cl-terminate T4 2.03 4.27 4.28 4.26 4.38 H3 1.99 4.08 4.14 4.18 7.35
下载: 导出CSV
-
[1] Bak S J, Mun D H, Jung K C, et al. 2013 Electron. Mater. Lett. 9 367
Google Scholar
[2] Semond F 2015 MRS Bull. 40 412
Google Scholar
[3] Liu X Y, Li H F, Uddin A, et al. 2007 J. Cryst. Growth 300 114
Google Scholar
[4] Cheng K, Leys M, Degroote S, et al. 2006 J. Electron. Mater. 35 592
Google Scholar
[5] Yuan Y, Zuo R, Mao K, et al. 2018 Appl. Surf. Sci. 436 50
Google Scholar
[6] Cordier Y, Comyn R, Frayssinet E, et al. 2018 Phys. Status Solidi A 215 1700637
Google Scholar
[7] Zhao D, Zhao D 2018 J. Semicond. 39 033006
Google Scholar
[8] Wang X, Li H, Wang J, et al. 2014 Electron. Mater. Lett. 10 1069
Google Scholar
[9] Liu B T, Ma P, Li X L, et al. 2017 Chin. Phys. Lett. 34 058101
Google Scholar
[10] Novák T, Kostelník P, Konečný M, et al. 2019 Jpn. J. Appl. Phys. 5 SC1018
[11] Lee S J, Jeon S R, Ju J W, et al. 2019 J. Nanosci. Nanotech. 19 892
Google Scholar
[12] Cao M S, Shu J C, Wang X X, et al. 2019 Ann. Phys. 531 1800390
Google Scholar
[13] Cao M S, Wang X X, Zhang M, et al. 2019 Adv. Funct. Mater. 29 1807398
Google Scholar
[14] Zhang M, Wang X X, Cao W Q, et al. 2019 Adv. Opt. Mater. 1900689
Google Scholar
[15] Albao M A, Hsu C H, Putungan D B, et al. 2010 Surf. Sci. 604 396
Google Scholar
[16] Luniakov Y V 2011 Surf. Sci. 605 1866
Google Scholar
[17] Matsuo Y, Kangawa Y, Togashi R, et al. 2007 J. Cryst. Growth 300 66
Google Scholar
[18] Kresse G 1993 Phys. Rev. B 47 558
Google Scholar
[19] Kresse G, Furthmüller J 1996 Phys. Rev. B 54 11169
Google Scholar
[20] Stadler R, Podloucky R, Kresse G, et al. 1998 Phys. Rev. B 57 4088
Google Scholar
[21] Henkelman G, Jónsson H 2000 J. Chem. Phys. 113 9978
Google Scholar
[22] Sheppard D, Terrell R, Henkelman G 2008 J. Chem. Phys. 128 134106
Google Scholar
[23] Sheppard D, Henkelman G 2011 J. Comput. Chem. 32 1769
Google Scholar
[24] Ly D Q, Paramonov L, Makatsoris C 2009 J. Phys.: Condens. Matter. 21 185006
Google Scholar
[25] Kepenekian M, Robles R, Rurali R, et al. 2014 Nanotechnology 25 465703
Google Scholar
[26] Hsieh M F, Lin D S, Tsay S F 2009 Phys. Rev. B 80 045304
Google Scholar

 下载:
下载:
计量
- 文章访问数: 15935
- PDF下载量: 227
- 被引次数: 0
