










-
采用基于密度泛函理论加U的计算方法, 研究了Ce和O空位单(共)掺杂锐钛矿相TiO2的电子结构和光吸收性质. 计算结果表明, Ce和O空位共掺杂TiO2的带隙中出现了杂质能级, 且带隙窄化为2.67 eV, 明显比纯TiO2和Ce, O空位单掺杂TiO2的要小, 因而可提高TiO2对可见光的响应能力, 使TiO2的光吸收范围增加. 光吸收谱显示, 掺杂后TiO2的光吸收边发生了显著红移; 在400.0—677.1 nm的可见光区, 共掺杂体系的光吸收强度显著高于纯TiO2和Ce单掺杂TiO2, 而略低于O空位单掺杂TiO2. 此外, Ce掺杂TiO2中引入O空位后, TiO2的导带边从−0.27 eV变化为−0.32 eV, 这表明TiO2的导带边的还原能力得到了加强. 计算结果为Ce和O空位共掺杂TiO2在可见光光解水方面的进一步研究提供了有力的理论依据.
The crystal structures, defect formation energy, electronic structures and optical properties of oxygen vacancy and/or Ce-(co)doped anatase TiO2 are investigated by using density functional theory plus U calculations. The calculated results indicate that lattice distortion induces the enhanced octahedral dipole moment in Ce doped TiO2 crystal when introducing oxygen vacancy into the lattice of the TiO2 crystal, which is effective for separating the photo-excited electron-hole pairs; meanwhile, compared with the valence band of pure TiO2 and TiO2 mono-doped separately with Ce and oxygen vacancy, the valence band of TiO2 co-doped with Ce and oxygen vacancy broadens drastically, which is mainly contributed from the electronic states of Ce 5d, Ti 4s and O 2p in the valence band shifting toward the lower energy direction. As a result, Ce doped TiO2 with oxygen vacancy is beneficial to the mobility of photo-generated carriers in TiO2. Similarly, the anti-bonding states also move toward the lower band energy direction, which are formed by the mixture of Ce 4f, Ce 5d, Ti 3d, and O 2p orbits in the conduction band. Due to these shifts, the energy gap of Ce and oxygen vacancy codoped TiO2 is narrowed to 2.67 eV with the emerge of the occupied impurity energy levels near Fermi level. Because of the above-mentioned excellence features, the absorption spectra for doped systems exhibit remarkable red-shift, especially, the intensity of optical absorption of TiO2 co-doped with Ce and oxygen vacancy in the visible region and the infra-red region are obviously stronger than those of the Ce mono-doped TiO2. When introducing oxygen vacancy into the Ce-doped system, the calculated conduction band energy edge position changes from −0.27 eV to −0.32 eV, which implies that the reducing power of the conduction band edge of TiO2 is remarkably enhanced. More fascinatingly, the calculated band energy edges for the Ce and oxygen vacancy codoped TiO2 can satisfy the basic requirement for water splitting under visible light irradiation. In conclusion, Ce and oxygen vacancy co-doped system can effectively strengthen the photo-catalytic activity of TiO2 and improve the utilization of the solar light; and our calculated results provide a powerful theoretical basis for the applications of the Ce and oxygen vacancy co-doped anatase TiO2 in visible-light-driven water splitting in the future research. -
Keywords:
- Ce doping /
- oxygen vacancy /
- anatase TiO2 /
- electronic structure /
- first-principles
[1] Fujishma A, Honda K 1972 Nature 238 37
 Google Scholar
Google Scholar
[2] Feng N D, Wang Q, Zheng A M, Zhang Z F, Fan J, Liu S B, Amoureux J P, Deng F 2013 J. Am. Chem. Soc. 135 1607
Google Scholar
[3] O’Regan B, Grätzel M 1991 Nature 3535 737
Google Scholar
[4] 封玉凤, 王利新 2013 玻璃与搪瓷 41 39
Google Scholar
Feng Y F, Wang L X 2013 Glass & Enamel 41 39
Google Scholar
[5] Bai H W, Liu L, Liu Z YM, Sun D D 2013 Water Res. 47 4126
Google Scholar
[6] Khan S U M, Al-Shahry M, Ingler W B 2002 Science 297 2243
Google Scholar
[7] Zhang J L, Wu Y M, Xing M Y, Leghari S A K, Sajjad S 2010 Energ. Environ. Sci. 3 715
Google Scholar
[8] Gai Y Q, Li J B, Li S S, Xia J B, Wei S H 2009 Phys. Rev. Lett. 102 036402
Google Scholar
[9] Asahi R, Morikawa T, Ohwaki T, Aoki K, Taga Y 2001 Science 293 269
Google Scholar
[10] Angelis F D, Vitillaro G, Kavan L, Nazeeruddin M K, Grätzel M 2012 J. Phys. Chem. C 116 18124
Google Scholar
[11] Mazierski P, Lisowski W, Grzyb T, Winiarski M J, Klimczuk T, Mikołajczyk A, Flisikowski J, Hirsch A, Kołakowska A, Puzyn T, Zaleska-Medynska A, Nadolna J 2017 Appl. Catal. B: Environ. 205 376
Google Scholar
[12] Du Y, Du M, Qiao Y, Dai J, Xu J, Yang P 2007 Colloid J. 69 695
Google Scholar
[13] Nasir M, Xi Z H, Xing M Y, Zhang J L, Chen F, Tian B Z, Bagwasi S 2013 J. Phys. Chem. C 117 9520
Google Scholar
[14] Li F B, Li X Z, Hou M F, Cheah K W, Choy W C H 2005 Appl. Catal. A: Gen. 285 181
Google Scholar
[15] Xiao G, Huang X, Liao X P, Shi B 2013 J. Phys. Chem. C 117 9739
Google Scholar
[16] Fu C, Li T Z, Qi J S, Pan J, Chen S H, Cheng C 2010 Chem. Phys. Lett. 494 117
Google Scholar
[17] Wei G D, Wei L, Chen Y X, Yan S S, Tian Y, Mei L G, Jiao J 2017 J. Alloy. Compd. 695 2261
Google Scholar
[18] 占昌朝, 许建, 曹小华, 徐冰洁, 李晨琪, 章志凯, 金文雄 2018 硅酸盐学报 46 564
Google Scholar
Zhan C C, Xu J, Cao X H, Xu B J, Li C Q, Zhang Z K, Jin W X 2018 J. Chin. Ceram. Soc. 46 564
Google Scholar
[19] Yang Y Q, Yin L C, Gong Y, Niu P, Wang J Q, Gu L, Chen X Q, Liu G, Wang L Z, Cheng H M 2018 Adv. Mater. 30 1704479
Google Scholar
[20] Long R, English N J 2009 J. Phys. Chem. C 113 8373
Google Scholar
[21] Zhou S W, Liu J, Peng P, Chen W Q 2015 Mod. Phys. Lett. B 29 1550249
Google Scholar
[22] Anisimov V I, Zaanen J, Andersen O K 1991 Phys. Rev. B 44 943
Google Scholar
[23] Han X P, Lee J C, Yoo H I 2009 Phys. Rev. B 79 100403(R)
Google Scholar
[24] Tang H, Berger H, Schmid P E, Lévy F, Burri G 1993 Solid State Commun. 87 847
Google Scholar
[25] Lenka M, Kamila K , Martin R, Libor C, Alice H, Pavlína P, Zdenek M, Lucie O, Anna W, PiotrK P, Andrzej K 2014 Appl. Catal. B: Environ. 152−153 172
Google Scholar
[26] Howard C J, Sabine T M, Dickson F 1991 Acta Crystallogr. Sec. B 47 462
Google Scholar
[27] Mo S D, Ching W Y 1995 Phys. Rev. B 51 13023
Google Scholar
[28] Burdett J K, Hughbanks T, Miller G J, Richardson J W J, Smith J V 1987 J. Am. Chem. Soc. 109 3639
Google Scholar
[29] 徐金荣, 王影, 朱兴凤, 李平, 张莉 2012 物理学报 20 207103
Google Scholar
Xu J R, Wang Y, Zhu X F, Li P, Zhang L 2012 Acta Phys. Sin. 20 207103
Google Scholar
[30] Sutassana N P, Sukit L, Jaejun Y 2017 Appl. Catal. B: Environ. 200 1
Google Scholar
[31] Zhang Y G, Wang Y X 2011 J. Appl. Phys. 110 033519
Google Scholar
[32] Anderson R A, Albert B, Ieda M G, Julio R S, Francesc I 2014 J. Phys. Chem. C 118 9677
Google Scholar
[33] Wu H C, Lin Y S, Lin S W 2013 Int. J. Photoenergy 2013 289328
Google Scholar
[34] Vinothkumar N, De M 2014 Mater. Renew. Sustain. Energy 3 25
Google Scholar
[35] Zhang H M, Yu X H, Mcleod J A, Sun X H 2014 Chem. Phys. Lett. 612 106
Google Scholar
[36] Rumaiz A K, Woicik J C, Cockayne E, Lin H Y, Jaffari H G, Shah S I 2009 Appl. Phys. Lett. 95 262111
Google Scholar
[37] 赵宗彦, 柳清菊, 朱忠其, 张瑾, 刘强 2008 功能材料 39 953
Google Scholar
Zhao Z Y, Liu Q J, Zhu Z Q, Zhang J, Liu Q 2008 J. Funct. Mater. 39 953
Google Scholar
[38] Tang J, Durrant J R, Klug D R 2008 J. Am. Chem. Soc. 130 13885
Google Scholar
[39] Linsebigler A L, Lu G Q, Yates J T 1995 Chem. Rev. 95 735
Google Scholar
[40] Liu S, Liu X P, Chen Y S, Jiang R Y 2010 J. Alloy. Compd. 506 877
Google Scholar
[41] Chen X B, Shen S H, Guo L J, Mao S S 2010 Chem. Rev. 110 6503
Google Scholar
[42] Brown G F, Wu J Q 2009 Laser Photon. Rev. 3 394
Google Scholar
[43] Sun J, Wang H T, He J L, Tian Y J 2005 Phys. Rev. B 71 125132
Google Scholar
[44] Yan N N, Zhu Z Q, Zhang J, Zhao Z Y, Liu Q J 2012 Mater. Res. Bull. 47 1869
Google Scholar
-
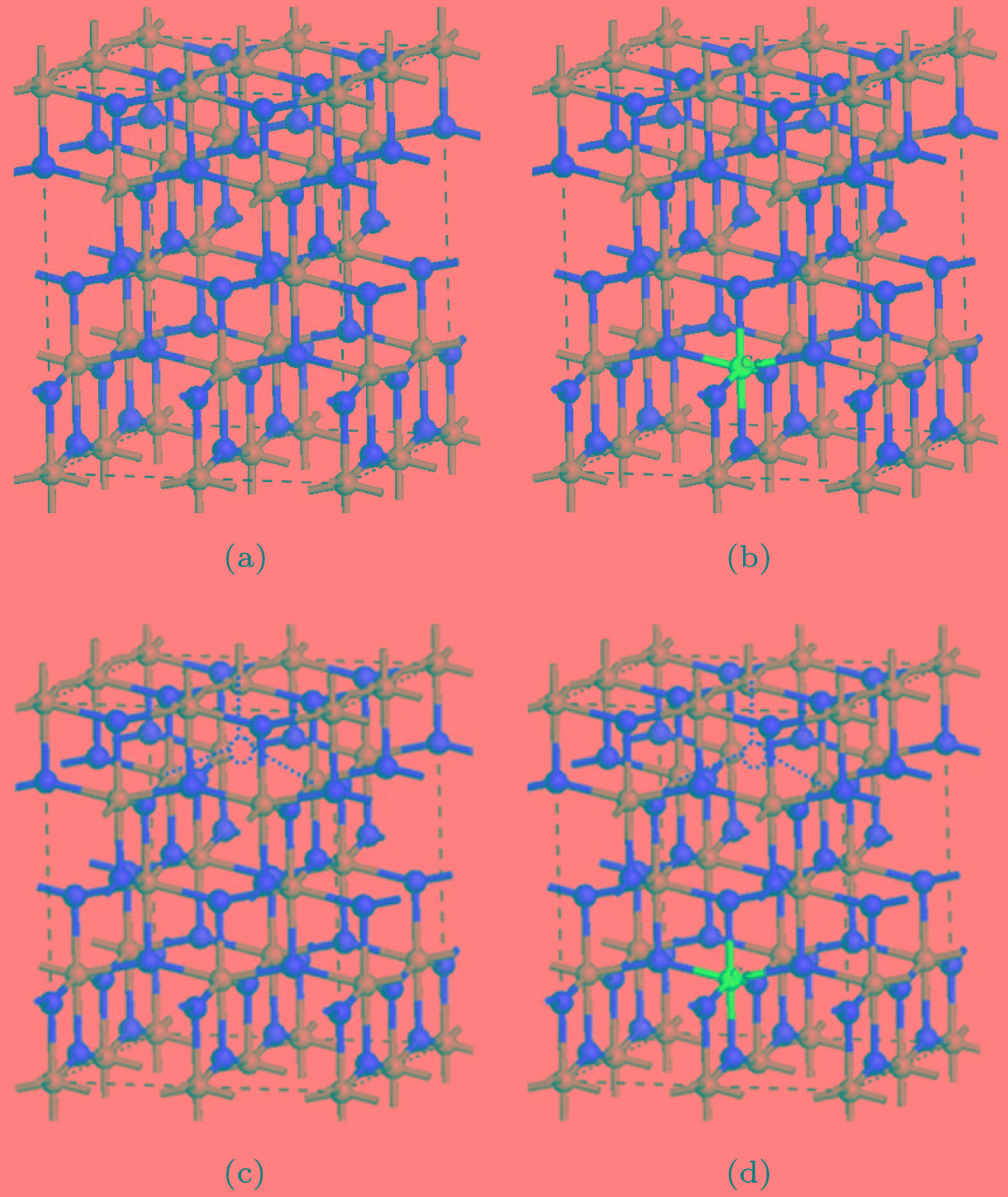
图 1 2 × 2 × 1的超胞结构图(红色球为O, 灰色球为Ti, 蓝色球表示Ce原子的掺杂位置, 红色虚圆圈表示OV的位置) (a) Pure TiO2; (b) Ce-TiO2; (c) OV-TiO2; (d) Ce/OV-TiO2
Fig. 1. The 2 × 2 × 1 supercell structure of various supercell models (the red, grey and blue balls represent O, Ti and Ce atoms, respectively; the imaginary red circle shows the position of oxygen vacancy): (a) Pure anatase TiO2; (b) Ce-TiO2; (c) OV-TiO2; (d) Ce/OV-TiO2.

图 2 纯TiO2和Ce/OV掺杂TiO2的能带结构图(图中虚线代表费米能级EF) (a) Pure TiO2; (b) Ce-TiO2; (c) OV-TiO2; (d) Ce/OV-TiO2
Fig. 2. Band structure of anatase TiO2 before and after doping (the dashed lines represent the Fermi level, EF): (a) Pure TiO2; (b) Ce-doped TiO2; (c) OV-TiO2; (d) Ce/OV-TiO2.
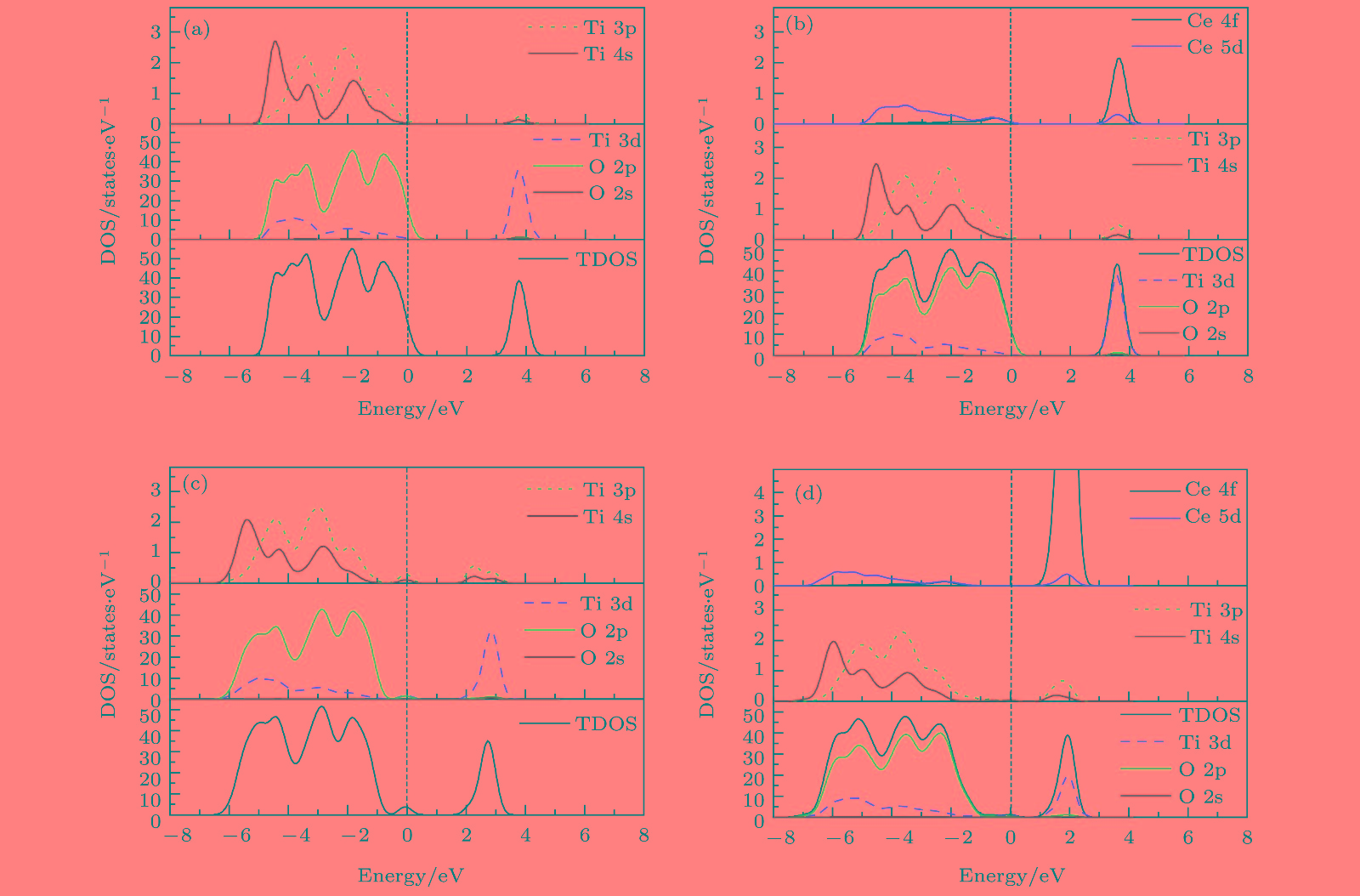
图 3 纯TiO2和Ce/OV掺杂TiO2的态密度图(图中虚线代表费米能级EF) (a) Pure TiO2; (b) Ce-TiO2; (c) OV-TiO2; (d) Ce/OV-TiO2
Fig. 3. Comparison of partial density of states of anatase TiO2 before and after doping (the dashed lines represent the Fermi level, EF): (a) Pure TiO2; (b) Ce-doped TiO2; (c) OV-TiO2; (d) Ce/OV-TiO2.

图 4 Ce/OV掺杂锐钛矿相TiO2的能带带边位置示意图(图中各带边位置的电位均以NHE的电位为参考零点, 粗黑线代表忽略杂质能级时的带边位置, 细红线代表考虑杂质能级时的带边位置)
Fig. 4. Calculated band energy positions of pure TiO2, Ce-TiO2, OV-TiO2, Ce/OV-TiO2 (the energy scale is indicated by the normal hydrogen electrode in electron volts as a reference; the short thick lines and fine red lines represent the band energy positions of TiO2 by neglecting and considering the impurity levels, respectively).
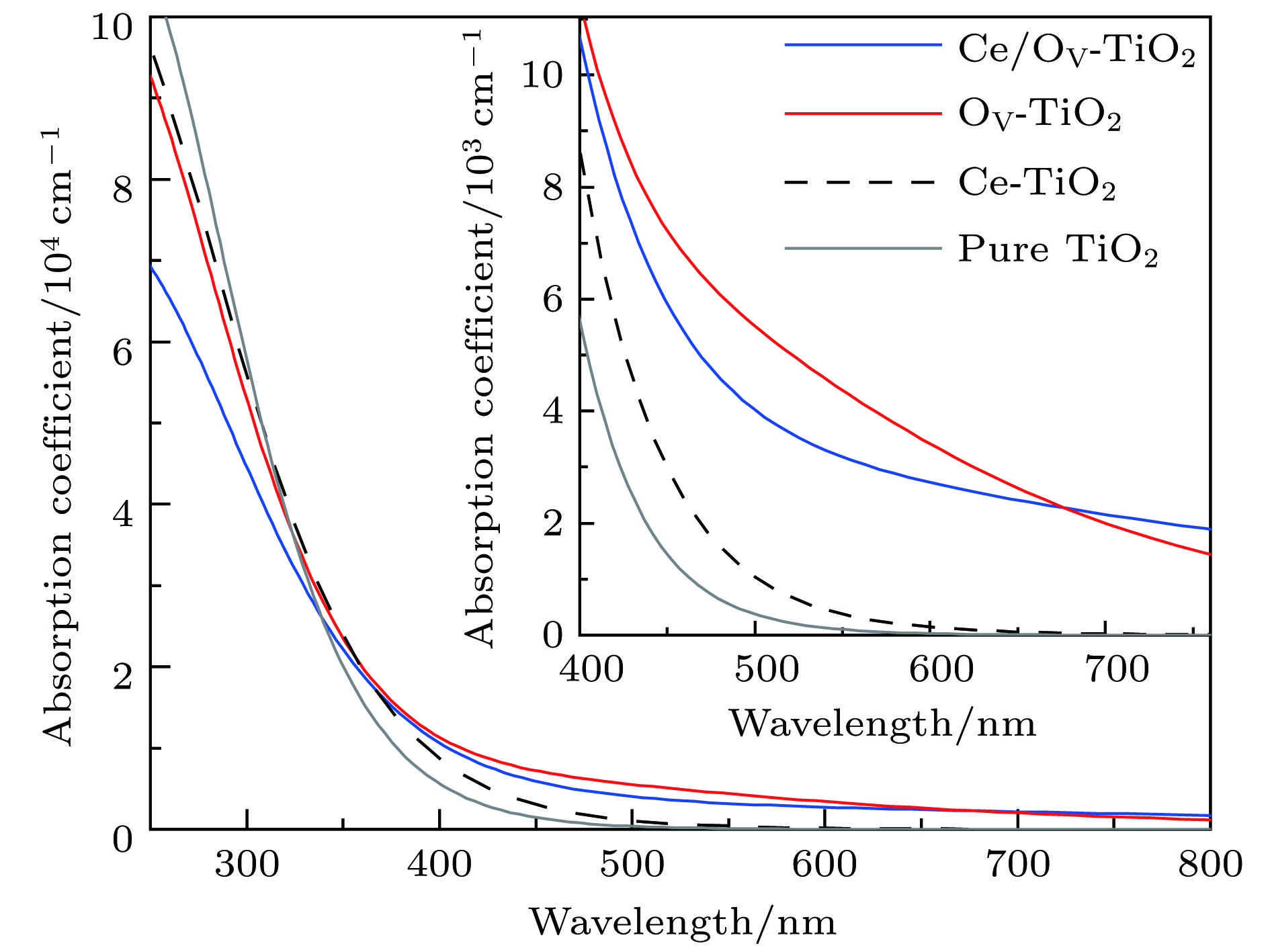
图 5 Ce/OV掺杂锐钛矿TiO2的光吸收谱(插图为可见光区光吸收谱的放大图)
Fig. 5. Absorption spectra of pure and different doping TiO2 models (inset is the expanded absorption spectra in the visible region).
表 1 采用GGA, GGA + U, LDA及LDA + U方法计算得到的纯锐钛矿相TiO2的晶格参数和带隙宽度
Table 1. Lattice parameters and band gap width of pure anatase TiO2 calculated at the GGA, GGA + U, LDA and LDA + U levels of theory.
参量 GGA GGA + U LDA LDA + U Experiment Theory Error% a/nm 0.3791 0.3897 0.3745 0.3806 0.3785[26] 0.3831[29] 0.5548 b/nm 0.3791 0.3897 0.3745 0.3806 0.3785[26] 0.3831[29] 0.5548 c/nm 0.9805 0.9925 0.9447 0.9674 0.9514[26] 0.9631[29] 1.6817 c/a 2.5860 2.5470 2.5230 2.5410 2.5140[26] 2.5140[29] 1.0739 u 0.2050 0.2050 0.2080 0.2070 — 0.2050[29] — dap/nm 0.2011 0.2031 0.1966 0.2005 0.1980[27] 0.1973[29] 1.2626 deq/nm 0.1946 0.2000 0.1914 0.1948 0.1934[28] 0.1930[29] 0.7238 Eg/eV 2.18 3.18 2.16 3.12 3.32[24] 3.23[29] — 注: dap, deq分别表示Ti—O长键和短键的键长; 误差指LDA + U方法相对于实验值的误差.  下载: 导出CSV
下载: 导出CSV
表 2 Ce/OV单(共)掺杂锐钛矿相TiO2弛豫后的晶胞参数
Table 2. Lattice parameters of Ce/OV single and co-doped anatase TiO2 after the structure relaxation.
参量 Ce-TiO2 OV-TiO2 Ce/OV-TiO2 This work Experiment Other theory This work This work a/nm 0.3822 0.3789[25] 0.3980[31] 0.3789 0.3827 b/nm 0.3833 — 0.3980[31] 0.3793 0.3798 c/nm 0.9956 0.9509[25] 0.9757[31] 0.9824 1.0090 dap/nm 0.2225 — 0.2273[32] 0.2012 0.2233 deq/nm 0.2183 — 0.2119[32] 0.1946 0.2252 注: dap, deq分别表示Ce-TiO2, Ce/OV-TiO2中Ce—O长键和短键的键长, 对于OV-TiO2则表示Ti—O长键和短键的平均键长.
下载: 导出CSV
表 3 Ce/OV掺杂锐钛矿相TiO2的缺陷形成能(Ef)、平均净电荷(q)和平均偶极矩(p)
Table 3. Defect formation energy, average net charges and average dipole moment of Ce/OV doped TiO2 supercell after the structure relaxation.
Model Ef/eV q/e p/Debye Ti-rich O-rich O Ti Ce Pure TiO2 — — −0.720 1.440 — 0.0000 Ce-TiO2 10.42 1.16 −0.719 1.405 1.920 0.0168 OV-TiO2 1.02 5.65 −0.727 1.412 — 0.0668 Ce/OV-TiO2 −15.84 −20.48 −0.725 1.373 1.920 0.0251
下载: 导出CSV
-
[1] Fujishma A, Honda K 1972 Nature 238 37
Google Scholar
[2] Feng N D, Wang Q, Zheng A M, Zhang Z F, Fan J, Liu S B, Amoureux J P, Deng F 2013 J. Am. Chem. Soc. 135 1607
Google Scholar
[3] O’Regan B, Grätzel M 1991 Nature 3535 737
Google Scholar
[4] 封玉凤, 王利新 2013 玻璃与搪瓷 41 39
Google Scholar
Feng Y F, Wang L X 2013 Glass & Enamel 41 39
Google Scholar
[5] Bai H W, Liu L, Liu Z YM, Sun D D 2013 Water Res. 47 4126
Google Scholar
[6] Khan S U M, Al-Shahry M, Ingler W B 2002 Science 297 2243
Google Scholar
[7] Zhang J L, Wu Y M, Xing M Y, Leghari S A K, Sajjad S 2010 Energ. Environ. Sci. 3 715
Google Scholar
[8] Gai Y Q, Li J B, Li S S, Xia J B, Wei S H 2009 Phys. Rev. Lett. 102 036402
Google Scholar
[9] Asahi R, Morikawa T, Ohwaki T, Aoki K, Taga Y 2001 Science 293 269
Google Scholar
[10] Angelis F D, Vitillaro G, Kavan L, Nazeeruddin M K, Grätzel M 2012 J. Phys. Chem. C 116 18124
Google Scholar
[11] Mazierski P, Lisowski W, Grzyb T, Winiarski M J, Klimczuk T, Mikołajczyk A, Flisikowski J, Hirsch A, Kołakowska A, Puzyn T, Zaleska-Medynska A, Nadolna J 2017 Appl. Catal. B: Environ. 205 376
Google Scholar
[12] Du Y, Du M, Qiao Y, Dai J, Xu J, Yang P 2007 Colloid J. 69 695
Google Scholar
[13] Nasir M, Xi Z H, Xing M Y, Zhang J L, Chen F, Tian B Z, Bagwasi S 2013 J. Phys. Chem. C 117 9520
Google Scholar
[14] Li F B, Li X Z, Hou M F, Cheah K W, Choy W C H 2005 Appl. Catal. A: Gen. 285 181
Google Scholar
[15] Xiao G, Huang X, Liao X P, Shi B 2013 J. Phys. Chem. C 117 9739
Google Scholar
[16] Fu C, Li T Z, Qi J S, Pan J, Chen S H, Cheng C 2010 Chem. Phys. Lett. 494 117
Google Scholar
[17] Wei G D, Wei L, Chen Y X, Yan S S, Tian Y, Mei L G, Jiao J 2017 J. Alloy. Compd. 695 2261
Google Scholar
[18] 占昌朝, 许建, 曹小华, 徐冰洁, 李晨琪, 章志凯, 金文雄 2018 硅酸盐学报 46 564
Google Scholar
Zhan C C, Xu J, Cao X H, Xu B J, Li C Q, Zhang Z K, Jin W X 2018 J. Chin. Ceram. Soc. 46 564
Google Scholar
[19] Yang Y Q, Yin L C, Gong Y, Niu P, Wang J Q, Gu L, Chen X Q, Liu G, Wang L Z, Cheng H M 2018 Adv. Mater. 30 1704479
Google Scholar
[20] Long R, English N J 2009 J. Phys. Chem. C 113 8373
Google Scholar
[21] Zhou S W, Liu J, Peng P, Chen W Q 2015 Mod. Phys. Lett. B 29 1550249
Google Scholar
[22] Anisimov V I, Zaanen J, Andersen O K 1991 Phys. Rev. B 44 943
Google Scholar
[23] Han X P, Lee J C, Yoo H I 2009 Phys. Rev. B 79 100403(R)
Google Scholar
[24] Tang H, Berger H, Schmid P E, Lévy F, Burri G 1993 Solid State Commun. 87 847
Google Scholar
[25] Lenka M, Kamila K , Martin R, Libor C, Alice H, Pavlína P, Zdenek M, Lucie O, Anna W, PiotrK P, Andrzej K 2014 Appl. Catal. B: Environ. 152−153 172
Google Scholar
[26] Howard C J, Sabine T M, Dickson F 1991 Acta Crystallogr. Sec. B 47 462
Google Scholar
[27] Mo S D, Ching W Y 1995 Phys. Rev. B 51 13023
Google Scholar
[28] Burdett J K, Hughbanks T, Miller G J, Richardson J W J, Smith J V 1987 J. Am. Chem. Soc. 109 3639
Google Scholar
[29] 徐金荣, 王影, 朱兴凤, 李平, 张莉 2012 物理学报 20 207103
Google Scholar
Xu J R, Wang Y, Zhu X F, Li P, Zhang L 2012 Acta Phys. Sin. 20 207103
Google Scholar
[30] Sutassana N P, Sukit L, Jaejun Y 2017 Appl. Catal. B: Environ. 200 1
Google Scholar
[31] Zhang Y G, Wang Y X 2011 J. Appl. Phys. 110 033519
Google Scholar
[32] Anderson R A, Albert B, Ieda M G, Julio R S, Francesc I 2014 J. Phys. Chem. C 118 9677
Google Scholar
[33] Wu H C, Lin Y S, Lin S W 2013 Int. J. Photoenergy 2013 289328
Google Scholar
[34] Vinothkumar N, De M 2014 Mater. Renew. Sustain. Energy 3 25
Google Scholar
[35] Zhang H M, Yu X H, Mcleod J A, Sun X H 2014 Chem. Phys. Lett. 612 106
Google Scholar
[36] Rumaiz A K, Woicik J C, Cockayne E, Lin H Y, Jaffari H G, Shah S I 2009 Appl. Phys. Lett. 95 262111
Google Scholar
[37] 赵宗彦, 柳清菊, 朱忠其, 张瑾, 刘强 2008 功能材料 39 953
Google Scholar
Zhao Z Y, Liu Q J, Zhu Z Q, Zhang J, Liu Q 2008 J. Funct. Mater. 39 953
Google Scholar
[38] Tang J, Durrant J R, Klug D R 2008 J. Am. Chem. Soc. 130 13885
Google Scholar
[39] Linsebigler A L, Lu G Q, Yates J T 1995 Chem. Rev. 95 735
Google Scholar
[40] Liu S, Liu X P, Chen Y S, Jiang R Y 2010 J. Alloy. Compd. 506 877
Google Scholar
[41] Chen X B, Shen S H, Guo L J, Mao S S 2010 Chem. Rev. 110 6503
Google Scholar
[42] Brown G F, Wu J Q 2009 Laser Photon. Rev. 3 394
Google Scholar
[43] Sun J, Wang H T, He J L, Tian Y J 2005 Phys. Rev. B 71 125132
Google Scholar
[44] Yan N N, Zhu Z Q, Zhang J, Zhao Z Y, Liu Q J 2012 Mater. Res. Bull. 47 1869
Google Scholar

 下载:
下载:
计量
- 文章访问数: 17520
- PDF下载量: 0
- 被引次数: 0
