










-
方镁石是镁方铁矿的终端组分, 化学组成为氧化镁(MgO). 理论预测的MgO熔化线和高压下实验测量结果之间存在巨大的分歧, 为澄清歧见人们展开了对MgO高压结构的进一步研究, 方镁石MgO高压结构预测及温度对结构稳定性的影响一直是高压凝聚态物理和地球物理领域的重要研究内容. 本文利用基于密度泛函理论的第一性原理计算方法, 对MgO实验结构、各种可能存在的结构及基于粒子群优化算法预测的晶体结构进行了系统深入的研究, 发现MgO在0—580 GPa的压力范围内一直以稳定岩盐结构存在, 580—800 GPa压力范围内的稳定结构为氯化铯结构. 尽管NiAs六角密堆结构和纤锌矿结构能合理解释冲击压缩实验中沿MgO的P-V雨贡纽线在(170 ± 10) GPa存在体积不连续的原因(Zhang L, Fei Y W 2008 Geophys. Res. Lett. 35 L13302)和高压下理论计算的熔化线与实验结果相差很大的原因(Aguado A, Madden P A 2005 Phys. Rev. Lett. 94 068501), 但这两种结构连同闪锌矿结构及基于粒子群优化算法预测的晶体结构B82和P3m1仅为其亚稳结构. 在MgO高压结构稳定性预测的基础上, 本文利用经典分子动力学方法, 分别引入能很好描述离子极化特性的壳层模型和离子压缩效应的呼吸壳层模型, 对MgO岩盐结构的高温稳定性进行了模拟研究, 给出了压力达150 GPa的高压熔化相图.Periclase is the terminal component of the ferropericlase, and its chemical composition is MgO. It is well known that there exists a huge difference between the melting curves of MgO determined experimentally and theoretically. A feasible way to clarify the nature of the melting temperature is to investigate the possible new phase of MgO. Meanwhile, it is very important to study the new phase and the influence of temperature on structural stability of MgO in high-pressure condensed matter physics and geophysics. In the present work, we study in detail the phase stability and the possible existing structures of MgO, which include the structure predicted by particle swarm optimization algorithm through using the first-principles pseudopotential density functional method. We find that MgO crystallizes into a rocksalt structure in a pressure range from 0 to 580 GPa and that the CsCl-type structure is of a high-pressure phase at up to 800 GPa. Although an NiAs-type hexagonal phase perhaps explains the volume discontinuity at (170 ± 10) GPa along the MgO Hugoniot in a shock-compression experiment (Zhang L, Fei Y W 2008 Geophys. Res. Lett. 35 L13302) and a wurtzite phase perhaps explains the huge difference between the melting curves of MgO determined experimentally and theoretically (Aguado A, Madden P A 2005 Phys. Rev. Lett. 94 068501), neither of them is existent in the entire range of pressures studied, according to the thermodynamic stability calculations. The calculations of phonon spectra indicate that the B3, B4, B81, B82, and P3m1 phases of MgO are dynamically stable at zero pressure. That is to say, all of the predicted structures are the metastable structures of MgO. In addition, the high-temperature structural stability of MgO is investigated by using very similar Lewis-Catlow and Stoneham-Sangster shell model potential based on the classical molecular dynamics (MD) simulations. In order to take into account the non-central force in crystal, the breathing shell model is also introduced in simulation. The thermodynamic melting curves are estimated on the basis of the thermal instability MD simulations and compared with the available experimental data and other theoretical results in the pressure range of 0-150 GPa.
-
Keywords:
- periclase /
- structural phase transition /
- melting /
- high pressure /
- high temperature
[1] Wood B J, Nell J 1991 Nature 351 309
 Google Scholar
Google Scholar
[2] Fei Y W 1999 Am. Meniral. 84 272
[3] Zhang L, Fei Y W 2008 Geophys. Res. Lett. 35 L13302
Google Scholar
[4] Francis M F, Taylor C D 2013 Phys. Rev. B 87 075450
Google Scholar
[5] Hong N V, Lan M T, Hung P K 2012 High Pressure Res. 32 509
Google Scholar
[6] Aguado A, Madden P A 2005 Phys. Rev. Lett. 94 068501
Google Scholar
[7] Yan Q, Rinke P, Winkelnkemper M, Qteish A, Bimberg D, Scheffler M, van de Walle C G 2012 Appl. Phys. Lett. 101 152105
Google Scholar
[8] Joshi K, Sharma B, Paliwal U, Barbiellini B 2012 J. Mater. Sci. 47 7549
Google Scholar
[9] Duffy T S, Hemley R J, Mao H K 1995 Phys. Rev. Lett. 74 1371
Google Scholar
[10] Song T, Sun X W, Liu Z J, Kong B, Quan W L, Fu Z J, Li J F, Tian J H 2012 Phys. Scr. 85 045702
Google Scholar
[11] Sims C E, Barrera G D, Allan N L, Mackrodt W C 1998 Phys. Rev. B 57 11164
Google Scholar
[12] Oganov A R, Gillan M J, Price G D 2003 J. Chem. Phys. 118 10174
Google Scholar
[13] Zerr A, Boehler R 1994 Nature 371 506
Google Scholar
[14] Anderson O L, Duba A 1997 J. Geophys. Res. 102 22659
Google Scholar
[15] Sun X W, Chen Q F, Chu Y D, Wang C W 2005 Physica B 370 186
Google Scholar
[16] Dubrovinsky L, Dubrovinskaia N, Prakapenka V B, Abakumov A M 2012 Nature Communs. 3 1163
Google Scholar
[17] Wang Y C, Lü J, Zhu L, Ma Y M 2010 Phys. Rev. B 82 094116
Google Scholar
[18] Lü J, Wang Y C, Zhu L, Ma Y M 2011 Phys. Rev. Lett. 106 015503
Google Scholar
[19] Li Y, Hao J, Liu H, Li Y, Ma Y 2014 J. Chem. Phys. 140 174712
Google Scholar
[20] Drozdov A P, Eremets M I, Troyan I A, Ksenofontov V, Shylin S I 2015 Nature 525 73
Google Scholar
[21] Peng F, Sun Y, Pickard C J, Needs R J, Wu Q, Ma Y 2017 Phys. Rev. Lett. 119 107001
Google Scholar
[22] Kresse G, Furthmüller J 1996 Phys. Rev. B 54 11169
Google Scholar
[23] Perdew J P, Burke K, Ernzerhof M 1996 Phys. Rev. Lett. 77 3865
Google Scholar
[24] Blöchl P E 1994 Phys. Rev. B 50 17953
Google Scholar
[25] Monkhorst H J, Pack J D 1976 Phys. Rev. B 13 5188
Google Scholar
[26] Segall M D, Lindan P J D, Probert M J, Pickard C J, Hasnip P J, Clark S J, Payne M C 2002 Phys. Condens. Matter. 14 2717
Google Scholar
[27] Perdew J P, Ruzsinszky A, Csonka G I, Vydrov O A, Scuseria G E, Constantin L A, Zhou X, Burke K 2008 Phys. Rev. Lett. 100 136406
Google Scholar
[28] Vanderbilt D 1990 Phys. Rev. B 41 7892
Google Scholar
[29] Fischer T H, Almlof J 1992 J. Phys. Chem. 96 9768
Google Scholar
[30] Gonze X, Lee C 1997 Phys. Rev B. 55 10355
Google Scholar
[31] Fincham D 1992 Mol. Sim. 8 165
Google Scholar
[32] Dick B G, Overhauser A W 1958 Phys. Rev. 112 90
Google Scholar
[33] Lewis G V, Catlow C R A 1985 J. Phys. C: Solid State Phys. 18 1149
Google Scholar
[34] Stoneham A M, Sangster M J L 1985 Phil. Mag. B 52 717
Google Scholar
[35] Catlow C R A, Faux I D, Norgett M J 1976 J. Phys. C: Solid State Phys. 9 419
[36] Henkelman G, Uberuaga B P, Harris D J, Harding J H, Allan N L 2005 Phys. Rev. B 72 115437
Google Scholar
[37] Mao H K, Bell P M 1979 J. Geophys. Res. 84 4533
Google Scholar
[38] Song T, Sun X W, Wang R F, Lu H W, Tian J H 2011 Physica B 406 293
Google Scholar
[39] Sun X W, Song T, Chu Y D, Liu Z J, Zhang Z R, Chen Q F 2010 Solid State Commun. 150 1785
Google Scholar
[40] Jeanloz R, Ahrens T J, Mao H K, Bell P M 1979 Science 206 829
Google Scholar
[41] Phillips J C 1971 Phys. Rev. Lett. 27 1197
Google Scholar
[42] van Camp P E, van Doren V E 1996 J. Phys.: Condens. Matter 8 3385
Google Scholar
[43] Cai Y X, Wu S T, Xu R, Yu J 2006 Phys. Rev. B 73 184104
Google Scholar
[44] Luo F, Cheng Y, Cai L C, Chen X R 2013 J. Appl. Phys. 113 033517
Google Scholar
[45] Speziale S, Zha C S, Duffy T S, Hemley R J, Mao H K 2001 J. Geophys. Res. 106 515
Google Scholar
[46] Lu K, Li Y 1998 Phys. Rev. Lett. 80 4474
Google Scholar
[47] Riley B 1966 Rev. Int. Hautes Temp. Refract. 3 327
[48] Tallon J L 1989 Nature 342 658
Google Scholar
[49] Belonoshko A B, Dubrovinsky L S 1996 Am. Mineral. 81 303
Google Scholar
[50] Wang Z, Tutti F, Saxena S K 2001 High Temp.-High Press. 33 357
Google Scholar
[51] Lindemann F A 1910 Z. Phys. 11 609
[52] Luo S N, Strachan A, Swift D C 2004 J. Chem. Phys. 120 11640
Google Scholar
-
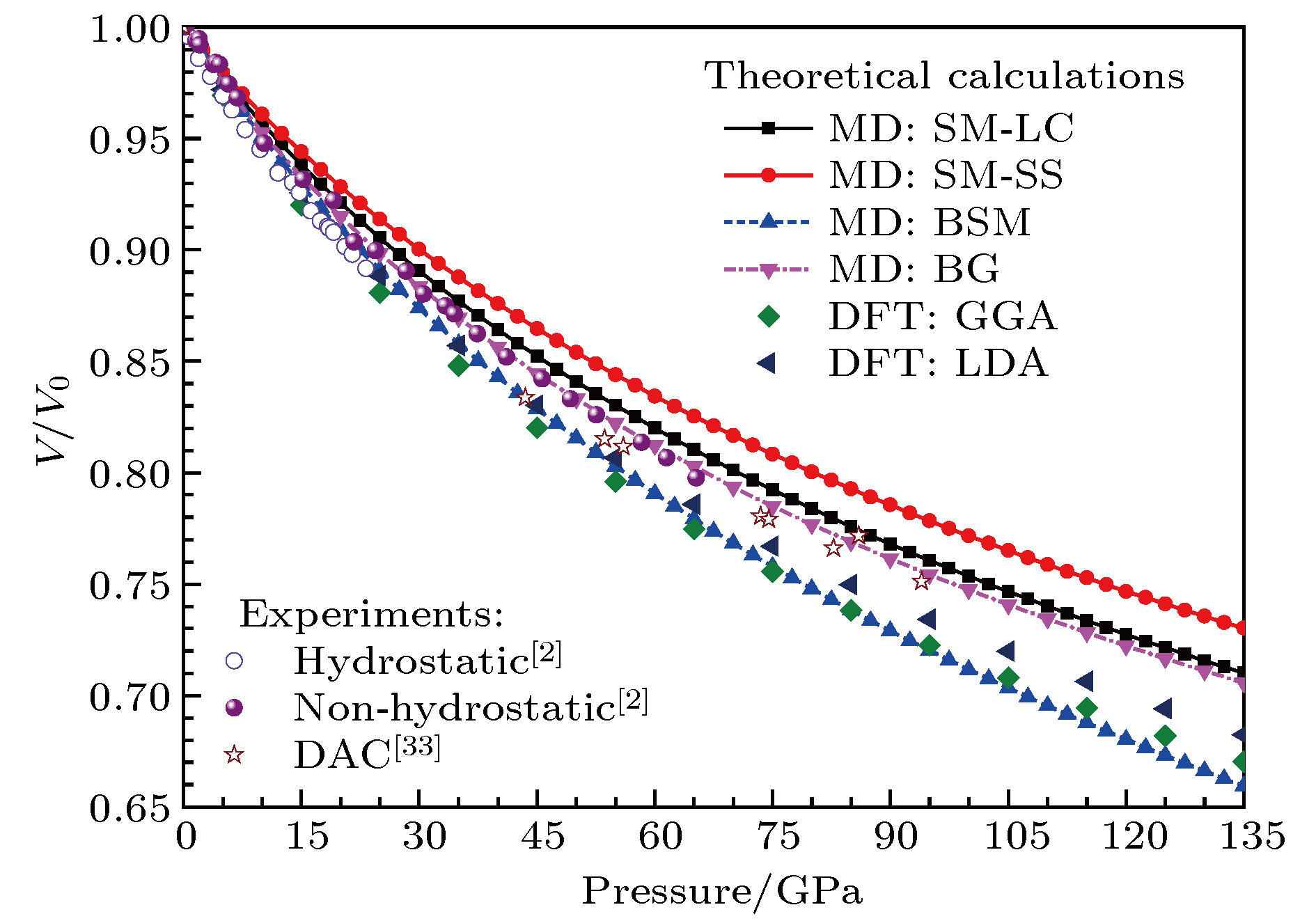
图 1 模拟得到的零温下MgO岩盐结构的体积比率随压力的变化及和实验结果的比较
Fig. 1. Volume ratios of MgO with NaCl-type structure under pressures at zero temperature, in comparison with the experimental data.

图 2 MgO晶体的可能结构(其中大球代表Mg原子, 小球代表O原子) (a) B1; (b) B2; (c) B3; (d) B4; (e) B81; (f) B82; (g) P3m1
Fig. 2. Probable crystal structures of MgO (the large and small spheres represent magnesium and oxygen atoms, respectively): (a) B1; (b) B2; (c) B3; (d) B4; (e) B81; (f) B82; (g) P3m1.
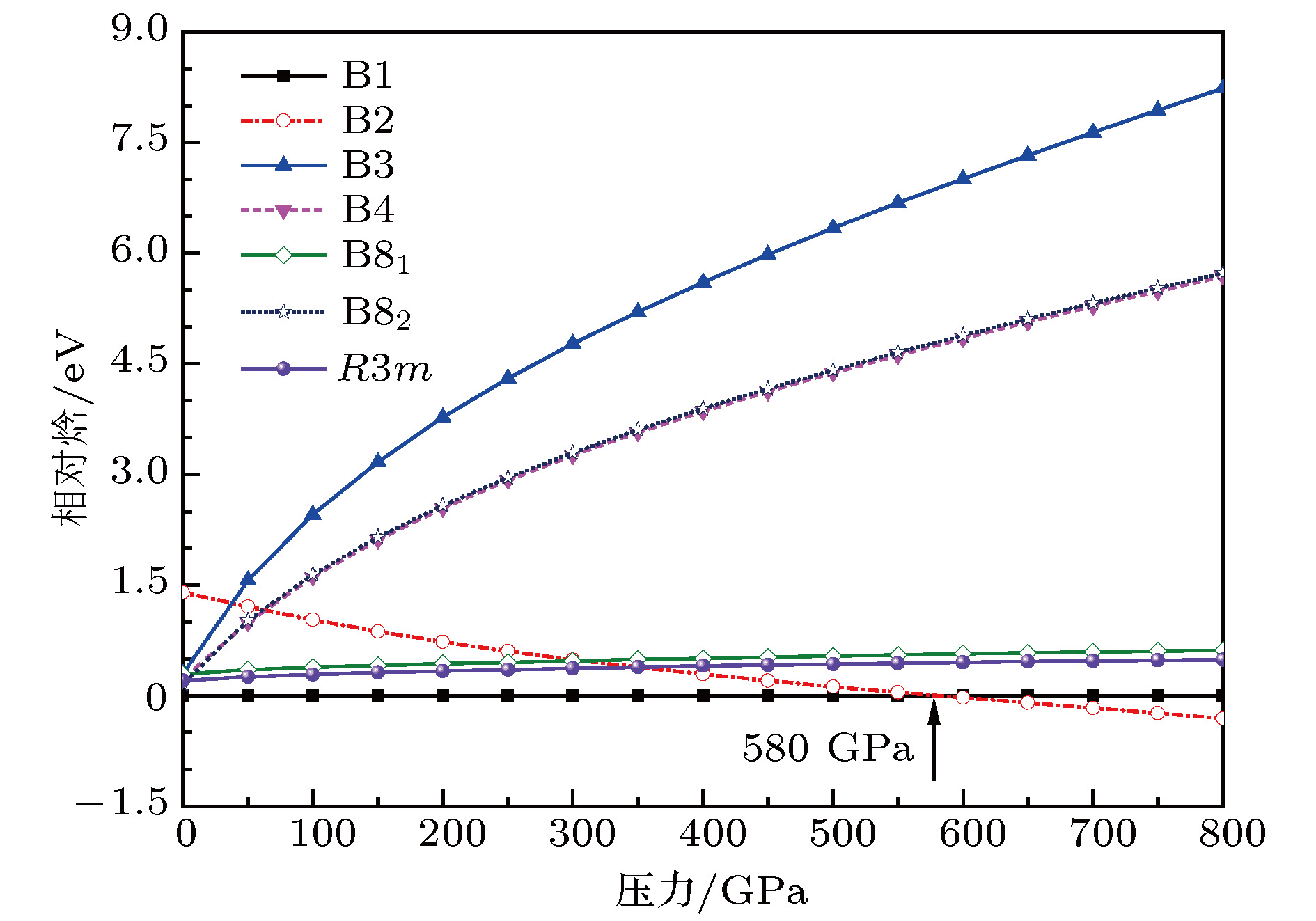
图 3 计算得到的MgO晶体在零温下的B1, B2, B3, B4, B81, B82和P3m1各可能结构的相对焓随压力的变化
Fig. 3. Calculated relative enthalpies of MgO with B1, B2, B3, B4, B81, B82 and P3m1 phases as a function of pressure at zero temperature.
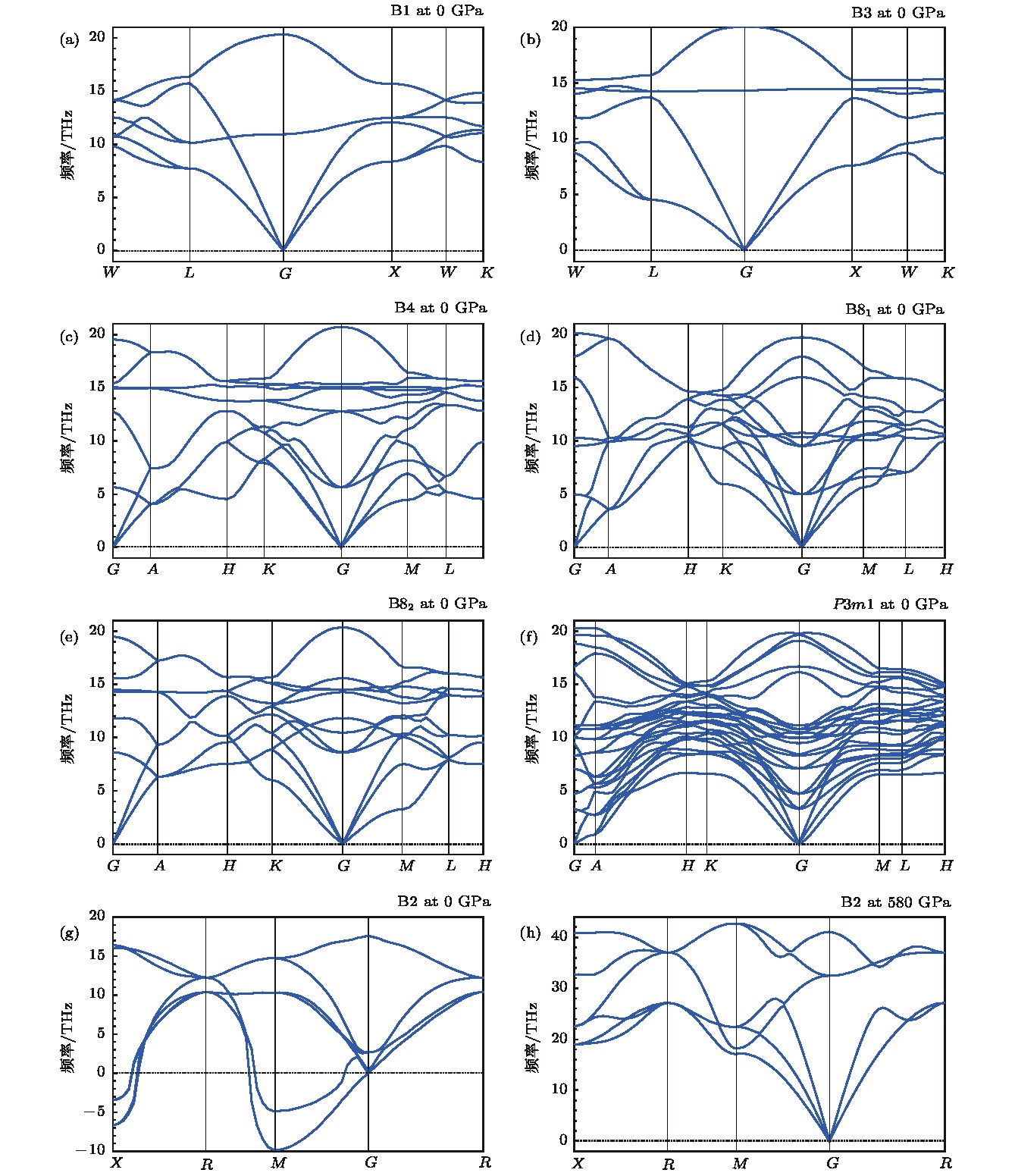
图 4 计算得到的MgO晶体B1 (a), B3 (b), B4 (c), B81 (d), B82 (e), P3m1 (f)和B2 (g)结构在零压下的声子谱和B2结构在相变压力为580 GPa下的声子谱(h)
Fig. 4. Calculated phonon spectra of MgO with B1 (a), B3 (b), B4 (c), B81 (d), B82 (e), P3m1 (f) and B2 (g) phases at 0 GPa and with B2 phase at 580 GPa (h).
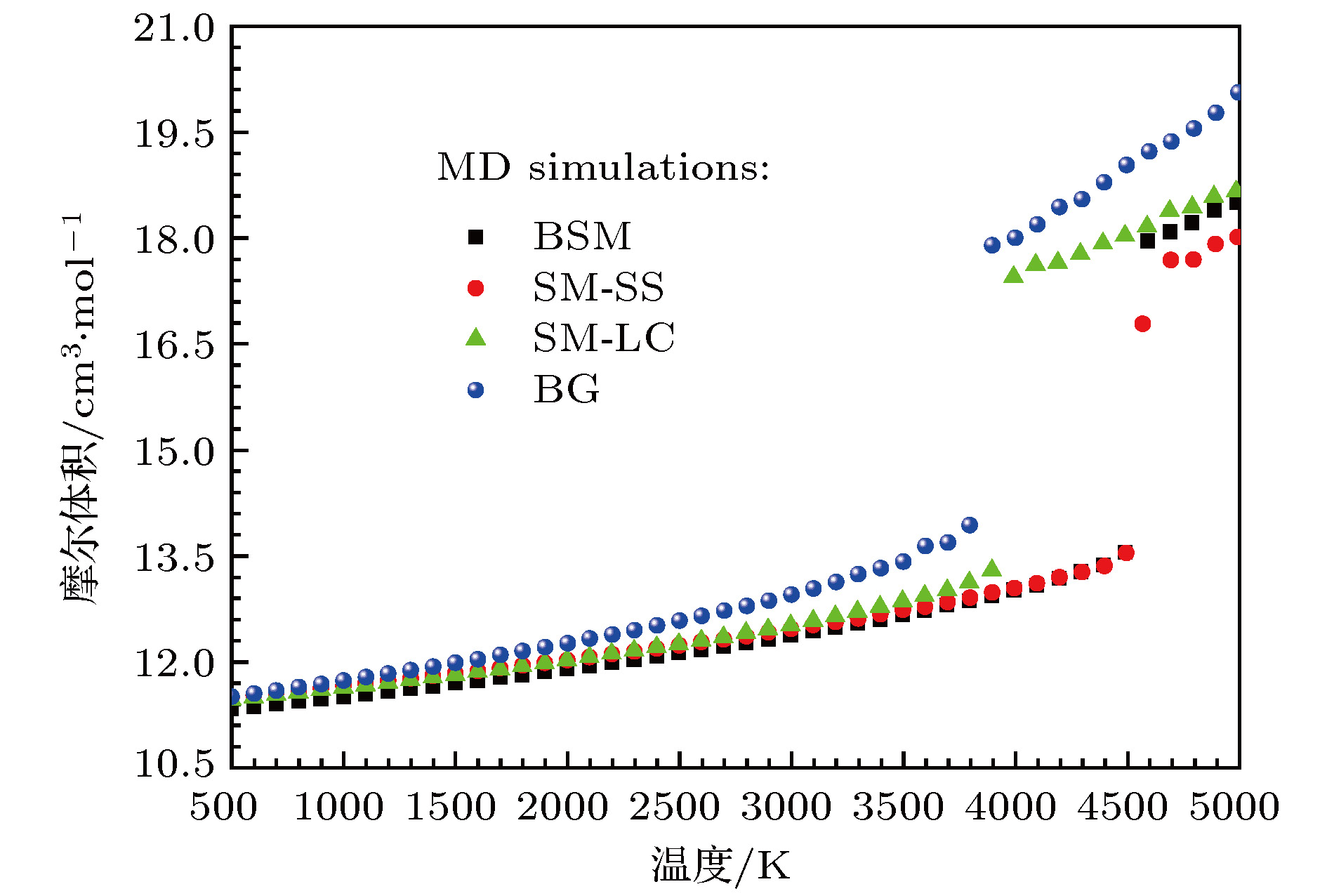
图 5 分子动力学模拟得到的零压下的MgO岩盐结构的体积随温度的变化
Fig. 5. Molecular dynamics calculated volume of MgO with NaCl-type structure as a function of temperature at zero pressure.
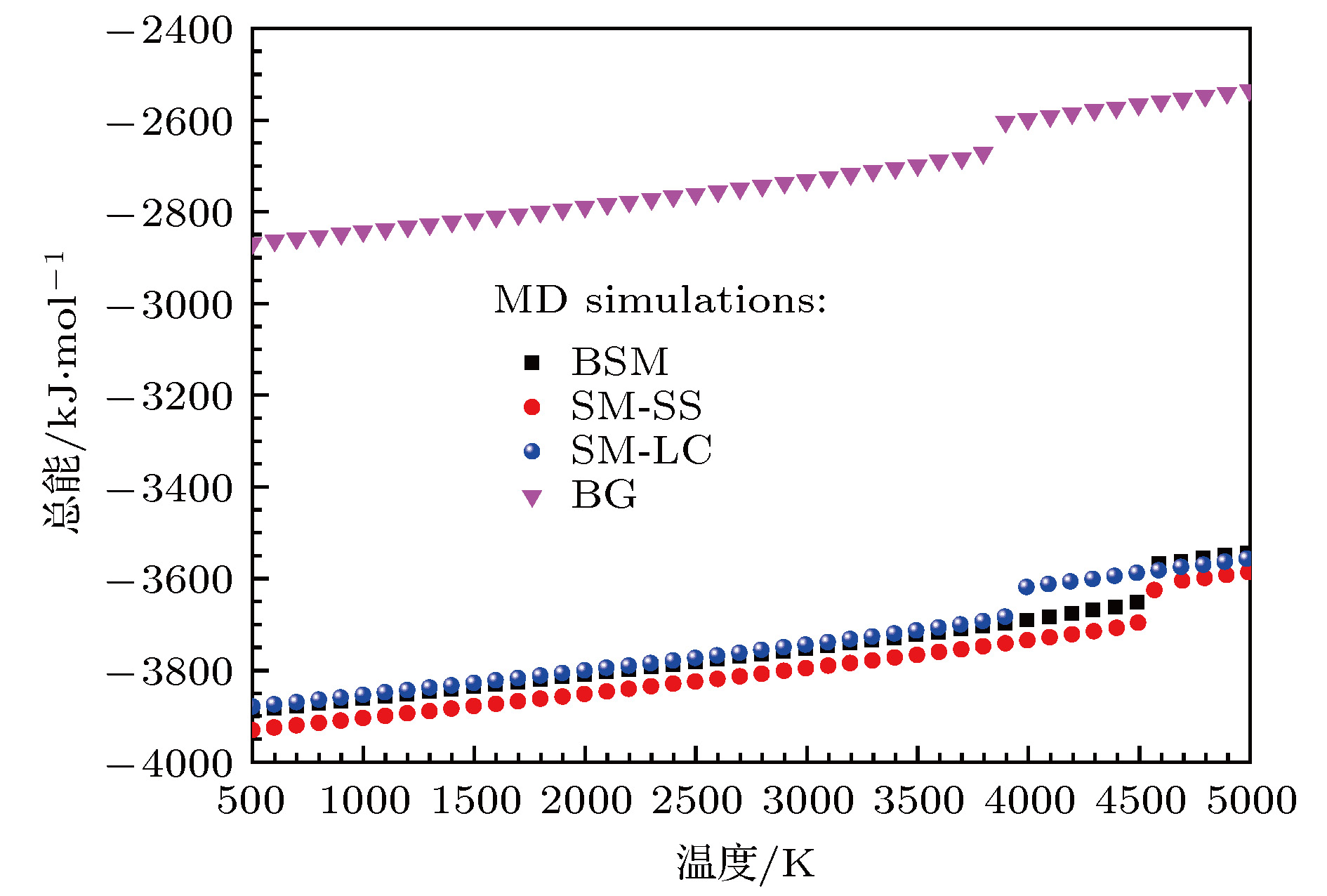
图 6 分子动力学模拟得到的零压下的MgO岩盐结构的总能随温度的变化
Fig. 6. Molecular dynamics calculated total energy of MgO with NaCl-type structure as a function of temperature at zero pressure.
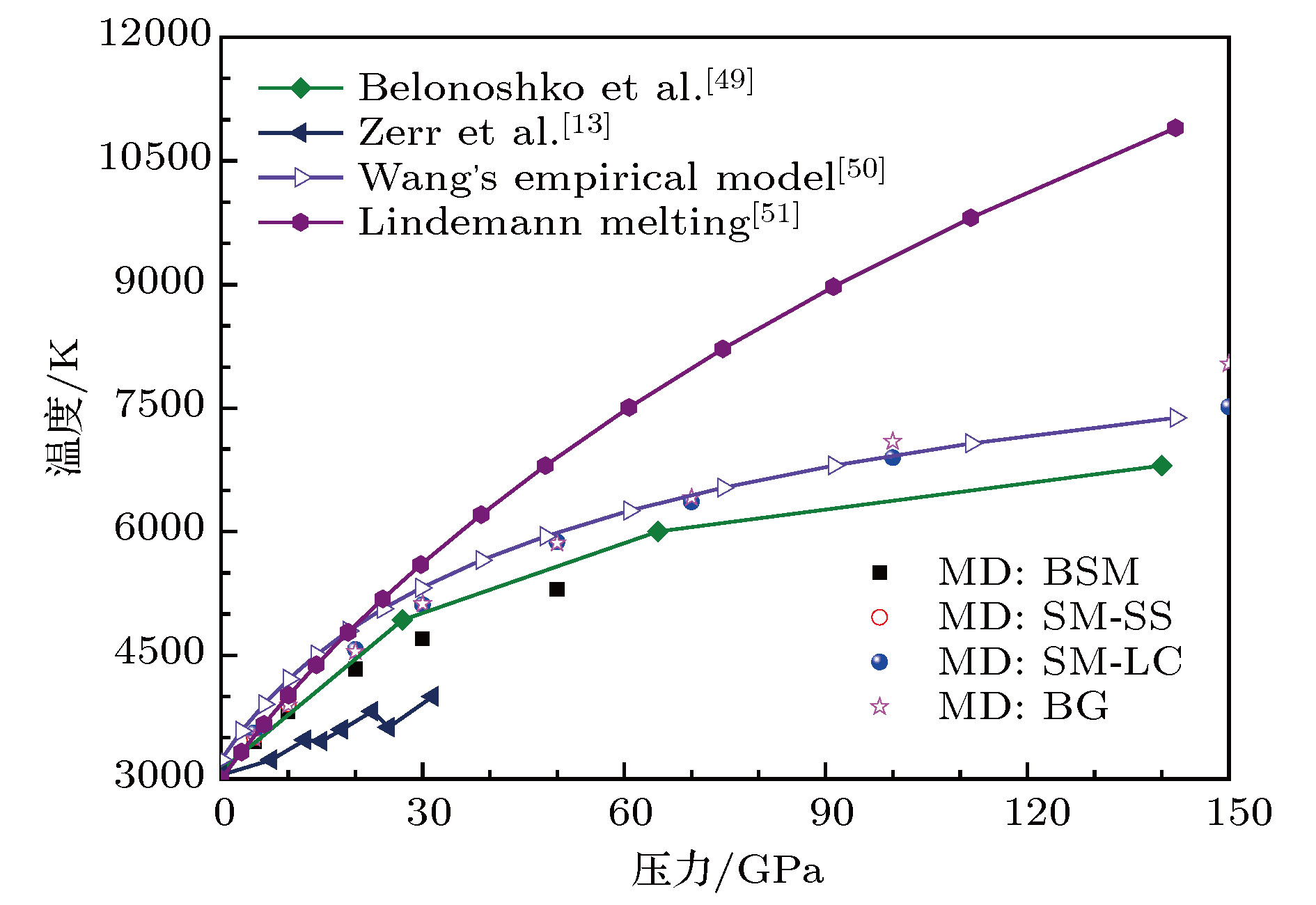
图 7 分子动力学模拟得到的MgO岩盐结构压力达150 GPa的熔化相图
Fig. 7. Molecular dynamics calculated melting phase diagram of MgO with NaCl-type structure as a function of pressure up to 150 GPa.
表 1 岩盐结构的MgO晶体特性模拟的短程对势参数
Table 1. Parameters of the short-range pair potentials for simulation of MgO crystal properties with NaCl-type structure.
Parameter SM-SS SM-LC BSM BG Mg-O Aij/eV 1346.6 821.6 1822.0 929.69 ρij/Å 0.2984 0.3243 0.32448 0.29909 Cij/eV·Å6 0.0 0.0 0.0 0.0 O-O Aij/eV 22764.4 22764.4 17.22556 4870.0 ρij/Å 0.149 0.149 0.65664 0.267 Cij/eV·Å6 20.37 20.37 1.1×10–7 77.0  下载: 导出CSV
下载: 导出CSV
表 2 岩盐结构的MgO晶体特性模拟的壳层及呼吸壳层参数
Table 2. Shell and breathing shell parameters of MgO crystal characteristic simulation with NaCl-type structure.
Parameter SM-SS SM-LC BSM Y/ |e| –2.9345 2.0000 –3.42274 k/ eV·Å–2 51.71 15.74 113.58 K/ eV·Å–2 298.304
下载: 导出CSV
-
[1] Wood B J, Nell J 1991 Nature 351 309
Google Scholar
[2] Fei Y W 1999 Am. Meniral. 84 272
[3] Zhang L, Fei Y W 2008 Geophys. Res. Lett. 35 L13302
Google Scholar
[4] Francis M F, Taylor C D 2013 Phys. Rev. B 87 075450
Google Scholar
[5] Hong N V, Lan M T, Hung P K 2012 High Pressure Res. 32 509
Google Scholar
[6] Aguado A, Madden P A 2005 Phys. Rev. Lett. 94 068501
Google Scholar
[7] Yan Q, Rinke P, Winkelnkemper M, Qteish A, Bimberg D, Scheffler M, van de Walle C G 2012 Appl. Phys. Lett. 101 152105
Google Scholar
[8] Joshi K, Sharma B, Paliwal U, Barbiellini B 2012 J. Mater. Sci. 47 7549
Google Scholar
[9] Duffy T S, Hemley R J, Mao H K 1995 Phys. Rev. Lett. 74 1371
Google Scholar
[10] Song T, Sun X W, Liu Z J, Kong B, Quan W L, Fu Z J, Li J F, Tian J H 2012 Phys. Scr. 85 045702
Google Scholar
[11] Sims C E, Barrera G D, Allan N L, Mackrodt W C 1998 Phys. Rev. B 57 11164
Google Scholar
[12] Oganov A R, Gillan M J, Price G D 2003 J. Chem. Phys. 118 10174
Google Scholar
[13] Zerr A, Boehler R 1994 Nature 371 506
Google Scholar
[14] Anderson O L, Duba A 1997 J. Geophys. Res. 102 22659
Google Scholar
[15] Sun X W, Chen Q F, Chu Y D, Wang C W 2005 Physica B 370 186
Google Scholar
[16] Dubrovinsky L, Dubrovinskaia N, Prakapenka V B, Abakumov A M 2012 Nature Communs. 3 1163
Google Scholar
[17] Wang Y C, Lü J, Zhu L, Ma Y M 2010 Phys. Rev. B 82 094116
Google Scholar
[18] Lü J, Wang Y C, Zhu L, Ma Y M 2011 Phys. Rev. Lett. 106 015503
Google Scholar
[19] Li Y, Hao J, Liu H, Li Y, Ma Y 2014 J. Chem. Phys. 140 174712
Google Scholar
[20] Drozdov A P, Eremets M I, Troyan I A, Ksenofontov V, Shylin S I 2015 Nature 525 73
Google Scholar
[21] Peng F, Sun Y, Pickard C J, Needs R J, Wu Q, Ma Y 2017 Phys. Rev. Lett. 119 107001
Google Scholar
[22] Kresse G, Furthmüller J 1996 Phys. Rev. B 54 11169
Google Scholar
[23] Perdew J P, Burke K, Ernzerhof M 1996 Phys. Rev. Lett. 77 3865
Google Scholar
[24] Blöchl P E 1994 Phys. Rev. B 50 17953
Google Scholar
[25] Monkhorst H J, Pack J D 1976 Phys. Rev. B 13 5188
Google Scholar
[26] Segall M D, Lindan P J D, Probert M J, Pickard C J, Hasnip P J, Clark S J, Payne M C 2002 Phys. Condens. Matter. 14 2717
Google Scholar
[27] Perdew J P, Ruzsinszky A, Csonka G I, Vydrov O A, Scuseria G E, Constantin L A, Zhou X, Burke K 2008 Phys. Rev. Lett. 100 136406
Google Scholar
[28] Vanderbilt D 1990 Phys. Rev. B 41 7892
Google Scholar
[29] Fischer T H, Almlof J 1992 J. Phys. Chem. 96 9768
Google Scholar
[30] Gonze X, Lee C 1997 Phys. Rev B. 55 10355
Google Scholar
[31] Fincham D 1992 Mol. Sim. 8 165
Google Scholar
[32] Dick B G, Overhauser A W 1958 Phys. Rev. 112 90
Google Scholar
[33] Lewis G V, Catlow C R A 1985 J. Phys. C: Solid State Phys. 18 1149
Google Scholar
[34] Stoneham A M, Sangster M J L 1985 Phil. Mag. B 52 717
Google Scholar
[35] Catlow C R A, Faux I D, Norgett M J 1976 J. Phys. C: Solid State Phys. 9 419
[36] Henkelman G, Uberuaga B P, Harris D J, Harding J H, Allan N L 2005 Phys. Rev. B 72 115437
Google Scholar
[37] Mao H K, Bell P M 1979 J. Geophys. Res. 84 4533
Google Scholar
[38] Song T, Sun X W, Wang R F, Lu H W, Tian J H 2011 Physica B 406 293
Google Scholar
[39] Sun X W, Song T, Chu Y D, Liu Z J, Zhang Z R, Chen Q F 2010 Solid State Commun. 150 1785
Google Scholar
[40] Jeanloz R, Ahrens T J, Mao H K, Bell P M 1979 Science 206 829
Google Scholar
[41] Phillips J C 1971 Phys. Rev. Lett. 27 1197
Google Scholar
[42] van Camp P E, van Doren V E 1996 J. Phys.: Condens. Matter 8 3385
Google Scholar
[43] Cai Y X, Wu S T, Xu R, Yu J 2006 Phys. Rev. B 73 184104
Google Scholar
[44] Luo F, Cheng Y, Cai L C, Chen X R 2013 J. Appl. Phys. 113 033517
Google Scholar
[45] Speziale S, Zha C S, Duffy T S, Hemley R J, Mao H K 2001 J. Geophys. Res. 106 515
Google Scholar
[46] Lu K, Li Y 1998 Phys. Rev. Lett. 80 4474
Google Scholar
[47] Riley B 1966 Rev. Int. Hautes Temp. Refract. 3 327
[48] Tallon J L 1989 Nature 342 658
Google Scholar
[49] Belonoshko A B, Dubrovinsky L S 1996 Am. Mineral. 81 303
Google Scholar
[50] Wang Z, Tutti F, Saxena S K 2001 High Temp.-High Press. 33 357
Google Scholar
[51] Lindemann F A 1910 Z. Phys. 11 609
[52] Luo S N, Strachan A, Swift D C 2004 J. Chem. Phys. 120 11640
Google Scholar

 下载:
下载:
计量
- 文章访问数: 25842
- PDF下载量: 168
- 被引次数: 0
