










-
胆红素是人体胆汁中的主要色素, 与人体健康密切相关. 结合荧光蛋白的胆红素分子代表一类新型荧光发色团, 在生物成像和生物传感领域有着重要应用. 本文结合隐式溶剂模型和线性响应含时密度泛函理论方法计算了胆红素分子最低单重激发态的垂直激发能、振子强度和垂直发射能. 以实验测量值和高水平RI-ADC(2)计算值作为参考, 系统考察了一系列密度泛函方法的预测表现, 结果发现最优化调控区间分离密度泛函方法整体表现最优, 预测的绝对误差和相对误差最小. 这得益于泛函中包含适宜的准确交换项比例能够产生既不离域也不局域的电子结构. 在最优化调控泛函方法计算的波函数基础上, 基于空穴-电子分析和片段间电荷转移方法定量表征了胆红素分子最低单重激发态, 发现其具有杂化局域-电荷转移的激发特征. 相信本工作可以为今后研究胆红素分子的激发态动力学过程和光谱性质提供重要理论依据, 该最优化调控理论模型也可以为接下来其他生物分子体系的激发态性质研究提供可靠、高效的理论工具.Bilirubin is the main pigment in human bile, which is closely related to human health. Bilirubin combining with fluorescent protein represents a new type of fluorescent chromophore and has important applications in the field of biological imaging and biosensor. Due to the lack of efficient and accurate electronic structure methods, the electronic structure and excited-state properties of bilirubin molecule are not characterized quantitatively and accurately. Firstly, the vertical absorption energy, oscillator strength and vertical emission energy of the lowest singlet excited state of bilirubin molecule are calculated by combining the implicit solvent model and the linear response time-dependent density functional theory (TDDFT) method. Compared to the experimental data and high-level RI-ADC(2) calculation, the prediction performance of a series of density functional methods is systematically investigated. The results show that the optimally-tuned range separated density functional method has the best overall performance and the minimum absolute and relative errors. This is obviously due to the fact that the suitable proportion of exact exchange included in density functionals can produce neither delocalized nor localized electronic structures. Based on the produced wavefunction by the optimally-tuned method, the excited-state characteristics of the S1 state of bilirubin molecule indicate a hybrid local and charge transfer excitation, based on the quantitative characterization using hole-electron analysis and interfragment charge transfer method. This work can provide a theoretical basis for the study of excited-state dynamics and spectral properties of bilirubin molecules and the optimally tuned range-separated DFT method also provide a reliable and efficient theoretical tool to study the excited-state properties of other biomolecular systems in the future.
-
Keywords:
- bilirubin /
- density functional theory /
- optimal tuning /
- excited-state character
[1] Stocker R, Yamamoto Y, Mcdonagh A F, Glazer A N, Ames B N 1987 Science 235 1043
 Google Scholar
Google Scholar
[2] Fevery J 2008 Liver Int. 28 592
Google Scholar
[3] Hissi E G V, Martinez J C G, Zamarbide G N, Estrada M R, Jensen S J K, Tomas-Vert F, Csizmadia I G 2009 J. Mol. Struc.: THEOCHEM 911 24
Google Scholar
[4] Carreira-Blanco C, Singer P, Diller R, Lustres J L P 2016 Phys. Chem. Chem. Phys. 18 7148
Google Scholar
[5] Person R V, Peterson B R, Lightner D A 1994 J. Am. Chem. Soc. 116 42
Google Scholar
[6] Nogales D, Lightner D A 1995 J. Biol. Chem. 270 73
Google Scholar
[7] Boiadjiev S E, Watters K, Wolf S, Lai B N, Welch W H, McDonagh A F, Lightner D A 2004 Biochemistry 43 15617
Google Scholar
[8] Lightner D A, Holmes D L, McDonagh A F 1996 J. Biol. Chem. 271 2397
Google Scholar
[9] Braslavsky S E, Holzwarth A R, Schaffner K 1983 Angew. Chem. Int. Ed. 22 656
Google Scholar
[10] Zietz B, Macpherson A N, Gillbro T 2004 Phys. Chem. Chem. Phys. 6 4535
Google Scholar
[11] Zietz B, Gillbro T 2007 J. Phys. Chem. B 111 11997
Google Scholar
[12] Cao X D, Zhang C C, Gao Z H, Liu Y Y, Zhao Y Z, Yang Y, Chen J Q, Jimenez R, Xu J H 2019 Phys. Chem. Chem. Phys. 21 2365
Google Scholar
[13] Zietz B, Blomgren F 2006 Chem. Phys. Lett. 420 556
Google Scholar
[14] Fabiano E, Della Sala F, Cingolani R, Weimer M, Görling A 2005 J. Phys. Chem. A 109 3078
Google Scholar
[15] Hammond J R, Kowalski K 2009 J. Chem. Phys. 130 194108
Google Scholar
[16] Budzák Š, Scalmani G, Jacquemin D 2017 J. Chem. Theory Comput. 13 6237
Google Scholar
[17] Hedin L 1965 Phys. Rev. 139 A796
Google Scholar
[18] Hybertsen M S, Louie S G 1986 Phys. Rev. B 34 5390
Google Scholar
[19] Kohn W, Sham L J 1965 Phys. Rev. 140 A1133
Google Scholar
[20] Cohen A J, Mori-Sanchez P, Yang W T 2011 Chem. Rev. 112 289
[21] Jacquemin D, Wathelet V, Perpète E A, Adamo C 2009 J. Chem. Theory Comput. 5 2420
Google Scholar
[22] Tian X H, Sun H T, Zhang Q S, Adachi C 2016 Chin. Chem. Lett. 27 1445
Google Scholar
[23] Sun H T, Autschbach J 2013 ChemPhysChem 14 2450
Google Scholar
[24] Jiang Y R, Hu Z B, Zhou B, Zhong C, Sun Z R, Sun H T 2019 J. Phys. Chem. C 123 5616
Google Scholar
[25] Sutton C, Sears J S, Coropceanu V, Brédas J 2013 J. Phys. Chem. Lett. 4 919
Google Scholar
[26] Stein T, Kronik L, Baer R 2009 J. Am. Chem. Soc. 131 2818
Google Scholar
[27] Sun H T, Zhong C, Brédas J 2015 J. Chem. Theory Comput. 11 3851
Google Scholar
[28] Penfold T J 2015 J. Phys. Chem. C 119 13535
Google Scholar
[29] Kronik L, Stein T, Refaely-Abramson S, Baer R 2012 J. Chem. Theory Comput. 8 1515
Google Scholar
[30] 孙海涛, 钟成, 孙真荣 2016 物理化学学报 32 2197
Google Scholar
Sun H T, Zhong C, Sun Z R 2016 Acta Phys.-Chim. Sin. 32 2197
Google Scholar
[31] Körzdörfer T, Sears J S, Sutton C, Brédas J 2011 J. Chem. Phys. 135 204107
Google Scholar
[32] Baer R, Livshits E, Salzner U 2010 Annu. Rev. Phys. Chem. 61 85
Google Scholar
[33] Stein T, Kronik L, Baer R 2009 J. Chem. Phys. 131 244119
Google Scholar
[34] Becke A D 1993 J. Chem. Phys. 98 5648
Google Scholar
[35] Lee C, Yang W T, Parr R G 1988 Phys. Rev. B 37 785
Google Scholar
[36] Grimme S, Ehrlich S, Goerigk L 2011 J. Comput. Chem. 32 1456
Google Scholar
[37] Ditchfield R, Hehre W J, Pople J A 1971 J. Chem. Phys. 54 724
Google Scholar
[38] Hehre W J, Ditchfield R, Pople J A 1972 J. Chem. Phys. 56 2257
Google Scholar
[39] Hariharan P C, Pople J A 1973 Theor. Chim. Acta 28 213
Google Scholar
[40] Perdew J P, Burke K, Ernzerhof M 1996 Phys. Rev. Lett. 77 3865
Google Scholar
[41] Yu H S, He X, Li S L, Truhlar D G 2016 Chem. Sci. 7 5032
Google Scholar
[42] Zhao Y, Truhlar D G 2008 Theor. Chem. Acc. 120 215
Google Scholar
[43] Zhao Y, Truhlar D G 2006 J. Phys. Chem. A 110 13126
Google Scholar
[44] Yanai T, Tew D P, Handy N C 2004 Chem. Phys. Lett. 393 51
Google Scholar
[45] Vydrov O A, Scuseria G E 2006 J. Chem. Phys. 125 234109
Google Scholar
[46] Chai J D, Head-Gordon M 2008 Phys. Chem. Chem. Phys. 10 6615
Google Scholar
[47] Peverati R, Truhlar D G 2011 J. Phys. Chem. Lett. 2 2810
Google Scholar
[48] Goerigk L, Grimme S 2010 J. Chem. Phys. 132 184103
Google Scholar
[49] Trofimov A B, Schirmer J 1995 J. Phys. B: At., Mol. Opt. Phys. 28 2299
Google Scholar
[50] Schäfer A, Horn H, Ahlrichs R 1992 J. Chem. Phys. 97 2571
Google Scholar
[51] Schäfer A, Huber C, Ahlrichs R 1994 J. Chem. Phys. 100 5829
Google Scholar
[52] Weigend F, Ahlrichs R 2005 Phys. Chem. Chem. Phys. 7 3297
Google Scholar
[53] Kumagai A, Ando R, Miyatake H, Greimel P, Kobayashi T, Hirabayashi Y, Shimogori T, Miyawaki A 2013 Cell 153 1602
Google Scholar
[54] Mennucci B 2012 Wiley Interdiscip. Rev.: Comput. Mol. Sci. 2 386
Google Scholar
[55] Tomasi J, Mennucci B, Cammi R 2005 Chem. Rev. 105 2999
Google Scholar
[56] Marenich A V, Cramer C J, Truhlar D G 2009 J. Phys. Chem. B 113 6378
Google Scholar
[57] Klamt A, Schüürmann G 1993 J. Chem. Soc., Perkin Trans. 2 799
[58] Frisch M J, Trucks G W, Schlegel H B, Scuseria G E, Robb M A, Cheeseman J R, Scalmani G, Barone V, Petersson G A, Nakatsuji H, Li X, Caricato M, Marenich A V, Bloino J, Janesko B G, Gomperts R, Mennucci B, Hratchian H P, Ortiz J V, Izmaylov A F, Sonnenberg J L, Williams, Ding F, Lipparini F, Egidi F, Goings J, Peng B, Petrone A, Henderson T, Ranasinghe D, Zakrzewski V G, Gao J, Rega N, Zheng G, Liang W, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Vreven T, Throssell K, Montgomery J J A, Peralta J E, Ogliaro F, Bearpark M J, Heyd J J, Brothers E N, Kudin K N, Staroverov V N, Keith T A, Kobayashi R, Normand J, Raghavachari K, Rendell A P, Burant J C, Iyengar S S, Tomasi J, Cossi M, Millam J M, Klene M, Adamo C, Cammi R, Ochterski J W, Martin R L, Morokuma K, Farkas O, Foresman J B, Fox D J 2013 Gaussian 09 Revision E. 01 Wallingford: Gaussian Inc
[59] Frisch M J, Trucks G W, Schlegel H B, Scuseria G E, Robb M A, Cheeseman J R, Scalmani G, Barone V, Petersson G A, Nakatsuji H, Li X, Caricato M, Marenich A V, Bloino J, Janesko B G, Gomperts R, Mennucci B, Hratchian H P, Ortiz J V, Izmaylov A F, Sonnenberg J L, Williams, Ding F, Lipparini F, Egidi F, Goings J, Peng B, Petrone A, Henderson T, Ranasinghe D, Zakrzewski V G, Gao J, Rega N, Zheng G, Liang W, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Vreven T, Throssell K, Montgomery J J A, Peralta J E, Ogliaro F, Bearpark M J, Heyd J J, Brothers E N, Kudin K N, Staroverov V N, Keith T A, Kobayashi R, Normand J, Raghavachari K, Rendell A P, Burant J C, Iyengar S S, Tomasi J, Cossi M, Millam J M, Klene M, Adamo C, Cammi R, Ochterski J W, Martin R L, Morokuma K, Farkas O, Foresman J B, Fox D J 2016 Gaussian 16 Revision A. 03 Wallingford: Gaussian Inc
[60] Neese F 2012 Wiley Interdiscip. Rev.: Comput. Mol. Sci. 2 73
Google Scholar
[61] Ahlrichs R, Bär M, Häser M, Horn H, Kölmel C 1989 Chem. Phys. Lett. 162 165
Google Scholar
[62] Lu T, Chen F W 2012 J. Comput. Chem. 33 580
Google Scholar
[63] Humphrey W, Dalke A, Schulten K 1996 J. Mol. Graphics 14 33
Google Scholar
-
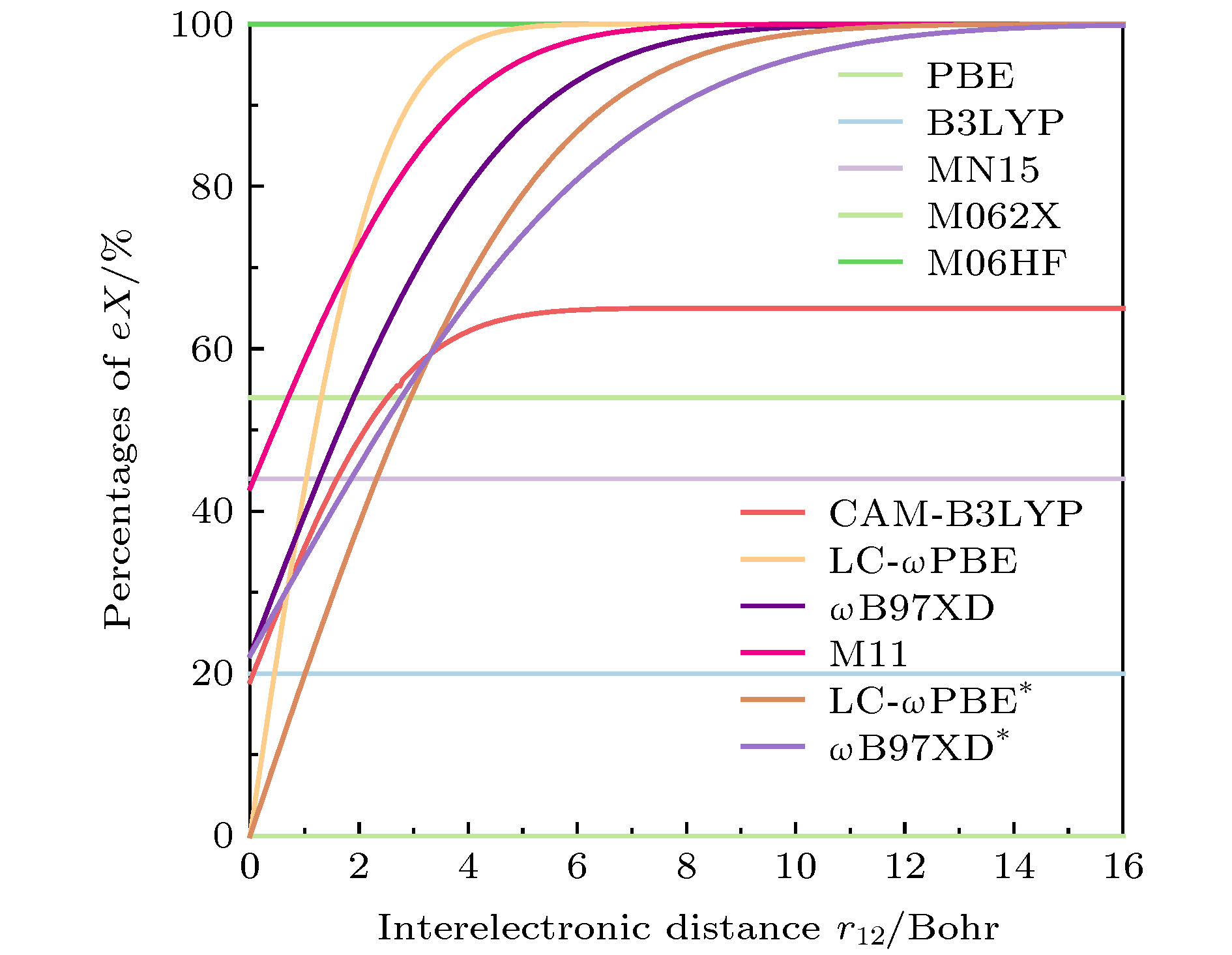
图 2 本文中各种密度泛函中所含准确交换项比例(eX%)与电子间距离(r12)关系示意图
Fig. 2. Percentages of exact-exchange (eX%) included in various density functionals as a function of intereletronic distance (r12, Bohr).

图 3 胆红素分子最低单重激发态的空穴-电子分布示意图
Fig. 3. Diagram of hole-electron distribution for the lowest singlet excited state (isovalue=0.001).
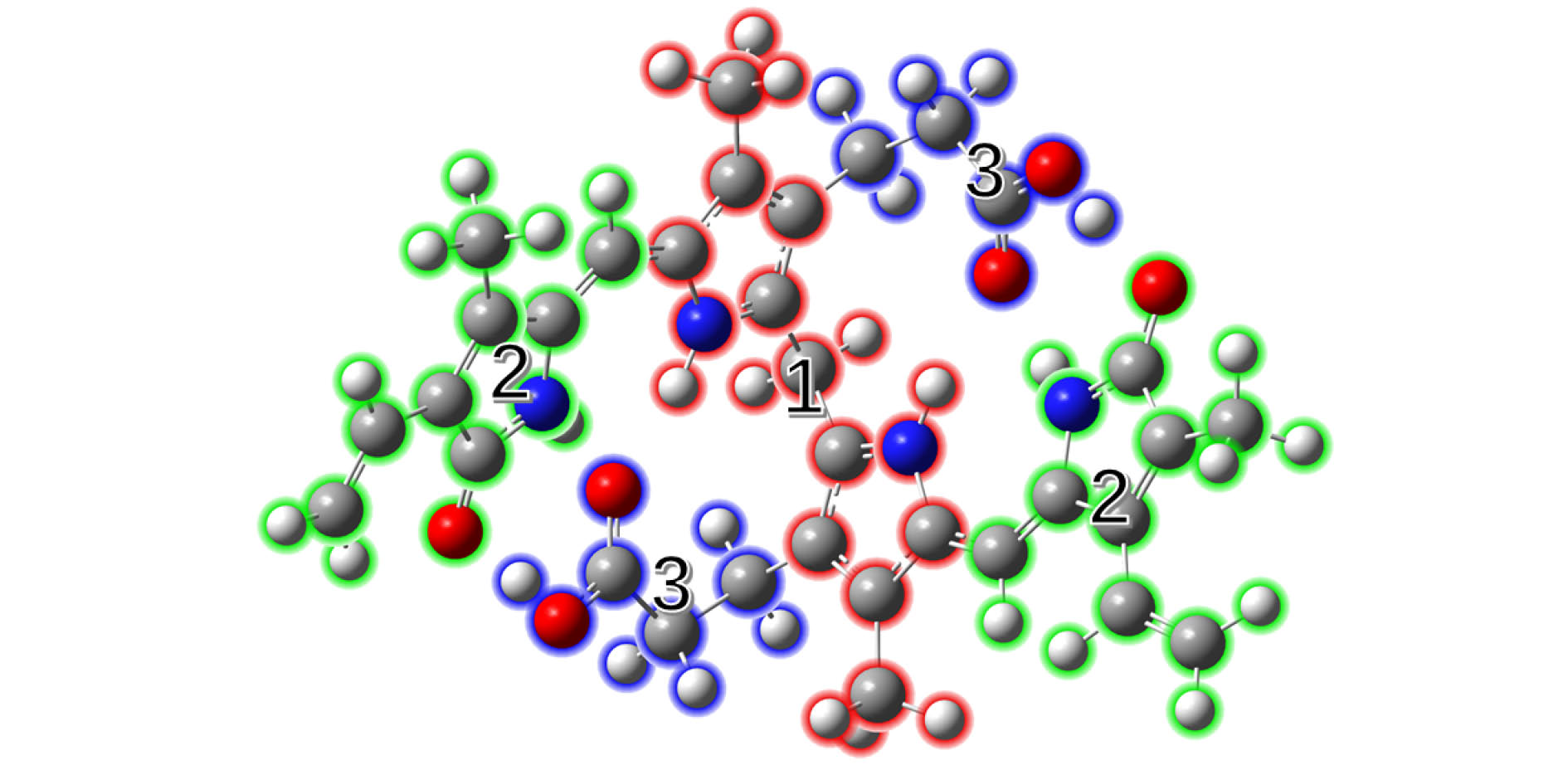
图 4 胆红素分子的三个片段划分及各片段对最低单重激发态的空穴和电子的贡献
Fig. 4. The divided three fragments of bilirubin molecule and contribution of each fragment to the hole and electron for the lowest singlet excited state.
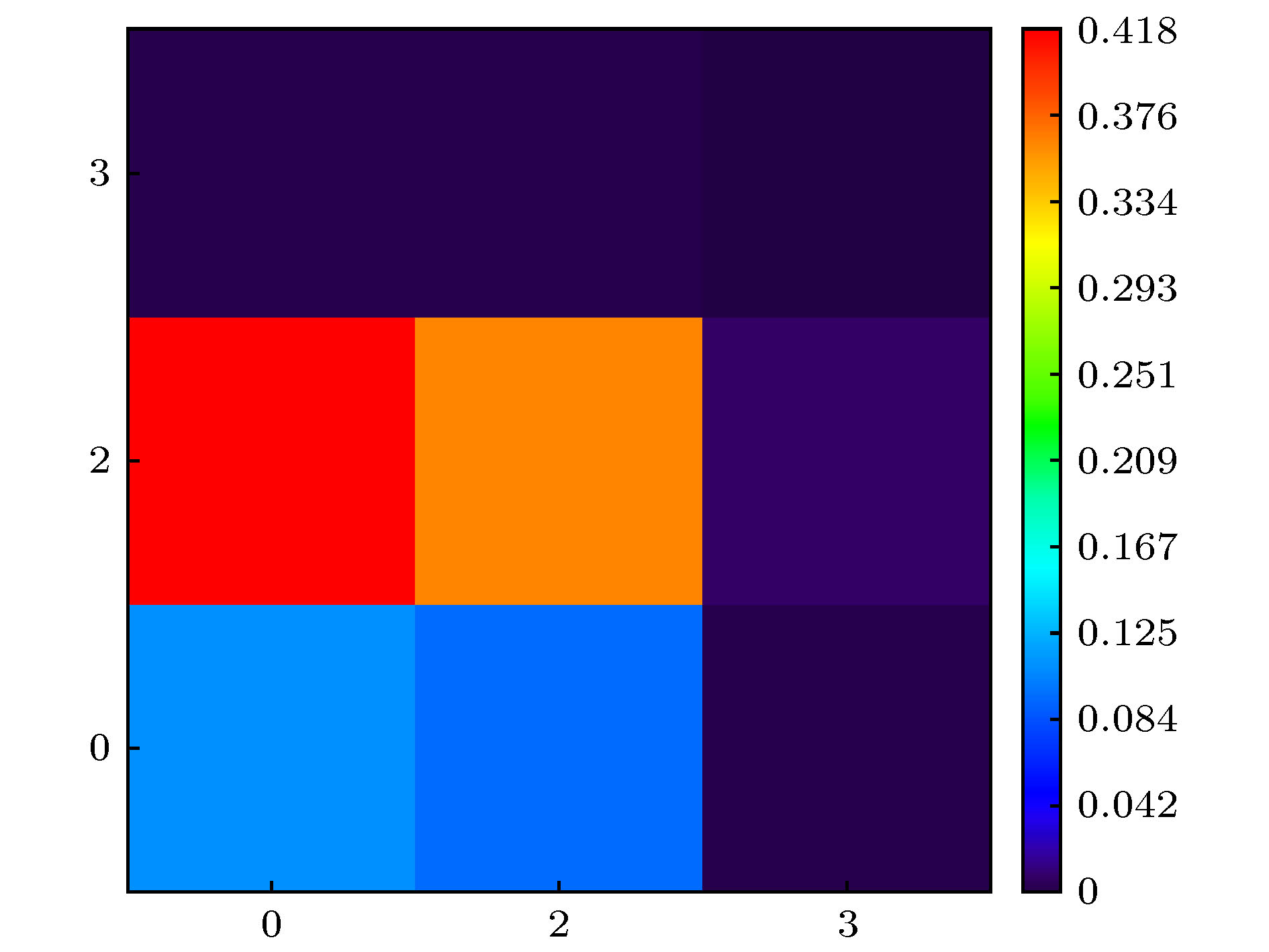
图 5 胆红素分子最低单重激发态的各片段电子转移矩阵热图
Fig. 5. Diagram of electron transfer matrix for each fragment of lowest singlet excited state of bilirubin molecule.
表 1 基组对计算的垂直激发能(EVA)的影响
Table 1. Influence of basis set on the calculated vertical excitation energy (EVA).
 下载: 导出CSV
下载: 导出CSV
表 2 各种理论方法计算胆红素分子的垂直激发能(EVA)、振子强度(f )和垂直发射能(EVE)以及与实验值相比的绝对误差和相对误差
Table 2. Vertical absorption energies (EVA), oscillator strength (f ) and vertical emission energies (EVE) of bilirubin and the absolute errors and relative errors compared to the available experimental data.
ω EVA/eV f (S1) AE/eV RE/% E VE/eV AE/eV RE/% PBE — 1.83 0.02 -0.90 33 1.68 -0.71 30 B3LYP — 2.48 0.12 -0.25 9 2.25 -0.14 6 MN15 — 2.81 1.21 0.08 3 2.40 0.01 0.4 M062X — 2.91 1.30 0.18 7 2.46 0.07 3 M06HF — 3.12 1.42 0.39 14 2.52 0.13 6 CAM-B3LYP 0.330 2.91 1.33 0.18 7 2.45 0.06 3 LC-ωPBE 0.400 3.11 1.43 0.38 14 2.54 0.15 6 ω B97XD 0.200 2.94 1.36 0.21 8 2.46 0.07 3 M11 0.250 3.01 1.40 0.28 10 2.49 0.10 4 LC-ω PBE* 0.178 2.76 1.20 0.03 1 2.36 -0.03 1 ω B97XD* 0.137 2.85 1.28 0.12 4 2.43 0.04 2 B2 GPPLYP — 2.92 1.13 0.19 7 — — — RI-ADC(2) — 2.69 1.06 -0.04 1 — — — EXP a — 2.73 — — — 2.39 — — a Experimental values are taken from Refs. [12,53].
下载: 导出CSV
表 3 胆红素分子最低单重激发态的各片段电子净变化量以及片段间电子转移量
Table 3. Net change of each fragment and electron transfer between fragments for lowest singlet excited state of bilirubin molecule.
Electron transfer between fragments 1 2 3 1 0.109 0.418 0.002 2 0.095 0.364 0.002 3 0.002 0.008 0.000 Net change of
each fragment–0.323 0.329 –0.006
下载: 导出CSV
-
[1] Stocker R, Yamamoto Y, Mcdonagh A F, Glazer A N, Ames B N 1987 Science 235 1043
Google Scholar
[2] Fevery J 2008 Liver Int. 28 592
Google Scholar
[3] Hissi E G V, Martinez J C G, Zamarbide G N, Estrada M R, Jensen S J K, Tomas-Vert F, Csizmadia I G 2009 J. Mol. Struc.: THEOCHEM 911 24
Google Scholar
[4] Carreira-Blanco C, Singer P, Diller R, Lustres J L P 2016 Phys. Chem. Chem. Phys. 18 7148
Google Scholar
[5] Person R V, Peterson B R, Lightner D A 1994 J. Am. Chem. Soc. 116 42
Google Scholar
[6] Nogales D, Lightner D A 1995 J. Biol. Chem. 270 73
Google Scholar
[7] Boiadjiev S E, Watters K, Wolf S, Lai B N, Welch W H, McDonagh A F, Lightner D A 2004 Biochemistry 43 15617
Google Scholar
[8] Lightner D A, Holmes D L, McDonagh A F 1996 J. Biol. Chem. 271 2397
Google Scholar
[9] Braslavsky S E, Holzwarth A R, Schaffner K 1983 Angew. Chem. Int. Ed. 22 656
Google Scholar
[10] Zietz B, Macpherson A N, Gillbro T 2004 Phys. Chem. Chem. Phys. 6 4535
Google Scholar
[11] Zietz B, Gillbro T 2007 J. Phys. Chem. B 111 11997
Google Scholar
[12] Cao X D, Zhang C C, Gao Z H, Liu Y Y, Zhao Y Z, Yang Y, Chen J Q, Jimenez R, Xu J H 2019 Phys. Chem. Chem. Phys. 21 2365
Google Scholar
[13] Zietz B, Blomgren F 2006 Chem. Phys. Lett. 420 556
Google Scholar
[14] Fabiano E, Della Sala F, Cingolani R, Weimer M, Görling A 2005 J. Phys. Chem. A 109 3078
Google Scholar
[15] Hammond J R, Kowalski K 2009 J. Chem. Phys. 130 194108
Google Scholar
[16] Budzák Š, Scalmani G, Jacquemin D 2017 J. Chem. Theory Comput. 13 6237
Google Scholar
[17] Hedin L 1965 Phys. Rev. 139 A796
Google Scholar
[18] Hybertsen M S, Louie S G 1986 Phys. Rev. B 34 5390
Google Scholar
[19] Kohn W, Sham L J 1965 Phys. Rev. 140 A1133
Google Scholar
[20] Cohen A J, Mori-Sanchez P, Yang W T 2011 Chem. Rev. 112 289
[21] Jacquemin D, Wathelet V, Perpète E A, Adamo C 2009 J. Chem. Theory Comput. 5 2420
Google Scholar
[22] Tian X H, Sun H T, Zhang Q S, Adachi C 2016 Chin. Chem. Lett. 27 1445
Google Scholar
[23] Sun H T, Autschbach J 2013 ChemPhysChem 14 2450
Google Scholar
[24] Jiang Y R, Hu Z B, Zhou B, Zhong C, Sun Z R, Sun H T 2019 J. Phys. Chem. C 123 5616
Google Scholar
[25] Sutton C, Sears J S, Coropceanu V, Brédas J 2013 J. Phys. Chem. Lett. 4 919
Google Scholar
[26] Stein T, Kronik L, Baer R 2009 J. Am. Chem. Soc. 131 2818
Google Scholar
[27] Sun H T, Zhong C, Brédas J 2015 J. Chem. Theory Comput. 11 3851
Google Scholar
[28] Penfold T J 2015 J. Phys. Chem. C 119 13535
Google Scholar
[29] Kronik L, Stein T, Refaely-Abramson S, Baer R 2012 J. Chem. Theory Comput. 8 1515
Google Scholar
[30] 孙海涛, 钟成, 孙真荣 2016 物理化学学报 32 2197
Google Scholar
Sun H T, Zhong C, Sun Z R 2016 Acta Phys.-Chim. Sin. 32 2197
Google Scholar
[31] Körzdörfer T, Sears J S, Sutton C, Brédas J 2011 J. Chem. Phys. 135 204107
Google Scholar
[32] Baer R, Livshits E, Salzner U 2010 Annu. Rev. Phys. Chem. 61 85
Google Scholar
[33] Stein T, Kronik L, Baer R 2009 J. Chem. Phys. 131 244119
Google Scholar
[34] Becke A D 1993 J. Chem. Phys. 98 5648
Google Scholar
[35] Lee C, Yang W T, Parr R G 1988 Phys. Rev. B 37 785
Google Scholar
[36] Grimme S, Ehrlich S, Goerigk L 2011 J. Comput. Chem. 32 1456
Google Scholar
[37] Ditchfield R, Hehre W J, Pople J A 1971 J. Chem. Phys. 54 724
Google Scholar
[38] Hehre W J, Ditchfield R, Pople J A 1972 J. Chem. Phys. 56 2257
Google Scholar
[39] Hariharan P C, Pople J A 1973 Theor. Chim. Acta 28 213
Google Scholar
[40] Perdew J P, Burke K, Ernzerhof M 1996 Phys. Rev. Lett. 77 3865
Google Scholar
[41] Yu H S, He X, Li S L, Truhlar D G 2016 Chem. Sci. 7 5032
Google Scholar
[42] Zhao Y, Truhlar D G 2008 Theor. Chem. Acc. 120 215
Google Scholar
[43] Zhao Y, Truhlar D G 2006 J. Phys. Chem. A 110 13126
Google Scholar
[44] Yanai T, Tew D P, Handy N C 2004 Chem. Phys. Lett. 393 51
Google Scholar
[45] Vydrov O A, Scuseria G E 2006 J. Chem. Phys. 125 234109
Google Scholar
[46] Chai J D, Head-Gordon M 2008 Phys. Chem. Chem. Phys. 10 6615
Google Scholar
[47] Peverati R, Truhlar D G 2011 J. Phys. Chem. Lett. 2 2810
Google Scholar
[48] Goerigk L, Grimme S 2010 J. Chem. Phys. 132 184103
Google Scholar
[49] Trofimov A B, Schirmer J 1995 J. Phys. B: At., Mol. Opt. Phys. 28 2299
Google Scholar
[50] Schäfer A, Horn H, Ahlrichs R 1992 J. Chem. Phys. 97 2571
Google Scholar
[51] Schäfer A, Huber C, Ahlrichs R 1994 J. Chem. Phys. 100 5829
Google Scholar
[52] Weigend F, Ahlrichs R 2005 Phys. Chem. Chem. Phys. 7 3297
Google Scholar
[53] Kumagai A, Ando R, Miyatake H, Greimel P, Kobayashi T, Hirabayashi Y, Shimogori T, Miyawaki A 2013 Cell 153 1602
Google Scholar
[54] Mennucci B 2012 Wiley Interdiscip. Rev.: Comput. Mol. Sci. 2 386
Google Scholar
[55] Tomasi J, Mennucci B, Cammi R 2005 Chem. Rev. 105 2999
Google Scholar
[56] Marenich A V, Cramer C J, Truhlar D G 2009 J. Phys. Chem. B 113 6378
Google Scholar
[57] Klamt A, Schüürmann G 1993 J. Chem. Soc., Perkin Trans. 2 799
[58] Frisch M J, Trucks G W, Schlegel H B, Scuseria G E, Robb M A, Cheeseman J R, Scalmani G, Barone V, Petersson G A, Nakatsuji H, Li X, Caricato M, Marenich A V, Bloino J, Janesko B G, Gomperts R, Mennucci B, Hratchian H P, Ortiz J V, Izmaylov A F, Sonnenberg J L, Williams, Ding F, Lipparini F, Egidi F, Goings J, Peng B, Petrone A, Henderson T, Ranasinghe D, Zakrzewski V G, Gao J, Rega N, Zheng G, Liang W, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Vreven T, Throssell K, Montgomery J J A, Peralta J E, Ogliaro F, Bearpark M J, Heyd J J, Brothers E N, Kudin K N, Staroverov V N, Keith T A, Kobayashi R, Normand J, Raghavachari K, Rendell A P, Burant J C, Iyengar S S, Tomasi J, Cossi M, Millam J M, Klene M, Adamo C, Cammi R, Ochterski J W, Martin R L, Morokuma K, Farkas O, Foresman J B, Fox D J 2013 Gaussian 09 Revision E. 01 Wallingford: Gaussian Inc
[59] Frisch M J, Trucks G W, Schlegel H B, Scuseria G E, Robb M A, Cheeseman J R, Scalmani G, Barone V, Petersson G A, Nakatsuji H, Li X, Caricato M, Marenich A V, Bloino J, Janesko B G, Gomperts R, Mennucci B, Hratchian H P, Ortiz J V, Izmaylov A F, Sonnenberg J L, Williams, Ding F, Lipparini F, Egidi F, Goings J, Peng B, Petrone A, Henderson T, Ranasinghe D, Zakrzewski V G, Gao J, Rega N, Zheng G, Liang W, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Vreven T, Throssell K, Montgomery J J A, Peralta J E, Ogliaro F, Bearpark M J, Heyd J J, Brothers E N, Kudin K N, Staroverov V N, Keith T A, Kobayashi R, Normand J, Raghavachari K, Rendell A P, Burant J C, Iyengar S S, Tomasi J, Cossi M, Millam J M, Klene M, Adamo C, Cammi R, Ochterski J W, Martin R L, Morokuma K, Farkas O, Foresman J B, Fox D J 2016 Gaussian 16 Revision A. 03 Wallingford: Gaussian Inc
[60] Neese F 2012 Wiley Interdiscip. Rev.: Comput. Mol. Sci. 2 73
Google Scholar
[61] Ahlrichs R, Bär M, Häser M, Horn H, Kölmel C 1989 Chem. Phys. Lett. 162 165
Google Scholar
[62] Lu T, Chen F W 2012 J. Comput. Chem. 33 580
Google Scholar
[63] Humphrey W, Dalke A, Schulten K 1996 J. Mol. Graphics 14 33
Google Scholar

 下载:
下载:
计量
- 文章访问数: 11250
- PDF下载量: 170
- 被引次数: 0
