










-
采用一种明显依赖于电子-电子距离的多参考组态相互作用方法, 结合ACVQZ基组进行了
${\rm CH}_2^+ $ 体系的从头算计算. 采用置换不变多项式神经网络方法对从头算得到的18222个单点能进行了拟合. 此外, 将势能面的特征与可得到的实验结果和理论结果进行了比较. 结果表明新构建的势能面比以往的势能面更加精确. 基于新构建的势能面, 采用准经典轨线方法进行了C+ + H2反应的动力学计算, 报道了积分截面和微分截面等动力学信息, 并与之前的理论结果进行了比较. 动力学结果表明在反应过程中插入反应机理占据主导地位.-
关键词:
- $ {\rm CH}_2^+ $体系 /
- 势能面 /
- 积分截面 /
- 微分截面
The multi-reference interaction method is explicitly dependent on the electron-electron distance, and ACVQZ basis set is used in the ab initio calculation. The potential energy surface (PES) is fitted by using the permutation invariant polynomial neural network method based on 18222 ab initio points. In addition, the topographical features of the PES are compared with available theoretical and experimental data. The results indicate that the present PES is more accurate and can be applied to any type of dynamic study. In order to validate the PES, the dynamic study of the C+ + H2 → H + CH+ reaction is carried out by using the quasi-classical trajectory method in a collision energy range of 0.4–1.0 eV. The integral cross sections and differential cross sections are calculated and compared with previous theoretical studies. For the integral cross section, the present results are, in general, in good agreement with previous theoretical studies, both of which increase with collision energy increasing. The forward and backward symmetric differential cross sections indicate that the “complex-forming” mechanism plays a dominant role in the reaction.-
Keywords:
- $ {\rm CH}_2^+ $ system /
- potential energy surface /
- integral cross section /
- differential cross section
[1] Douglas A E, Herzberg G 1941 Astrophys. J. 94 381
 Google Scholar
Google Scholar
[2] Hierl P M, Morris R A, Viggiano A A 1997 J. Chem. Phys. 106 10145
Google Scholar
[3] Plasil R, Mehner T, Dohnal P, Kotrik T, Glosik J, Gerlich D 2011 Astrophys. J. 737 60
Google Scholar
[4] Armentrout P B 2000 Int. J. Mass Spect. 200 219
Google Scholar
[5] Luca A, Borodi G, Gerlich D 2006 Photonic, Electronic and Atomic Collisions (Singapore: World Scientific)
[6] Federer W, Villinger H, Howorka F, Lindinger W, Tosi P, Bassi D, Ferguson E 1984 Phys. Rev. Lett. 52 2084
Google Scholar
[7] Stoecklin T, Halvick P 2005 Phys. Chem. Chem. Phys. 7 2446
Google Scholar
[8] Halvick P, Stoecklin T, Larrégaray P, Bonnet L 2007 Phys. Chem. Chem. Phys. 9 582
Google Scholar
[9] Zanchet A, Godard B, Bulut N, Roncero O, Halvick P, Cernicharo J 2013 Astrophys. J. 766 80
Google Scholar
[10] Warmbier R, Schneider R 2011 Phys. Chem. Chem. Phys. 13 10285
Google Scholar
[11] Li Y Q, Zhang P Y, Han K L 2015 J. Chem. Phys. 142 124302
Google Scholar
[12] Guo J, Zhang A J, Zhou Y, Liu J Y, Jia J F, Wu H S 2017 Chem. Phys. Lett. 689 121
Google Scholar
[13] Sundaram P, Manivannan V, Padamanadan R 2017 Phys. Chem. Chem. Phys. 19 20172
Google Scholar
[14] Sundaram P, Padamanadan R 2018 J. Chem. Phys. 148 164306
Google Scholar
[15] Sundaram P, Padamanadan R 2020 J. Phys. B-At. Mol. Opt. 53 105201
Google Scholar
[16] Wu H, Duan Z, Chen G 2020 Chem. Phys. Lett. 755 137783
Google Scholar
[17] Guo L, Ma H Y, Zhang L L, Song Y Z, Li Y Q 2018 RSC Adv. 8 13635
Google Scholar
[18] Herráez-Aguilar D, Jambrina P G, et al. 2014 Phys. Chem. Chem. Phys. 16 24800
Google Scholar
[19] Werfelli G, Halvick P, Honvault P, Kerkeni B, Stoecklin T 2015 J. Chem. Phys. 143 114304
Google Scholar
[20] Bovino S, Grassi T, Gianturco F A 2015 J. Phys. Chem. A 119 11973
Google Scholar
[21] Faure A, Halvick P, Stoecklin T, et al. 2017 Mon. Not. R. Astron. Soc. 469 612
Google Scholar
[22] Guo L, Yang Y F, Fan X X, Ma F C, Li Y Q 2017 Commun. Theor. Phys. 67 549
Google Scholar
[23] Gerlich D, Horning S 1992 Chem. Rev. 92 1509
Google Scholar
[24] Gerlich D, Borodi G, Luca A, Mogo C, Smith M 2011 Z. Phys. Chem. 225 475
Google Scholar
[25] Biglari Z, Shayesteh A, Maghari A 2014 Comput. Theor. Chem. 1047 22
Google Scholar
[26] Reddy R R, Nazeer A Y, Rama G K, Baba B D 2004 J. Quant. Spectrosc. Radiat. Transfer 85 105
Google Scholar
[27] Varandas A J C 1996 J. Chem. Phys. 105 3524
Google Scholar
[28] Song Y Z, Zhang Y, Zhang L L, Gao S B, Meng Q T 2015 Chin. Phys. B 24 063101
Google Scholar
[29] Yang C L, Huang Y J, Zhang X, Han K L 2003 J. Mol. Struct.: Theochem. 625 289
Google Scholar
[30] May A J, Valeev E F, Polly R, Manby F R 2005 Phys. Chem. Chem. Phys. 7 2710
Google Scholar
[31] Dunning T H 1989 J. Chem. Phys. 90 1007
Google Scholar
[32] Kendall R A, Dunning T H, Harrison R J 1992 J. Chem. Phys. 96 6796
Google Scholar
[33] Werner H J, Knowles P J 1985 J. Chem. Phys. 82 5053
Google Scholar
[34] Knowles P J, Werner H J 1985 Chem. Phys. Lett. 115 259
Google Scholar
[35] Jiang B, Li J, Guo H 2016 Int. Rev. Phys. Chem. 35 479
Google Scholar
[36] Jiang B, Guo H 2013 J. Chem. Phys. 139 054112
Google Scholar
[37] Li W T, He D, Sun Z G 2019 J. Chem. Phys. 151 185102
Google Scholar
[38] Li W T, Wang X M, Zhao H L, He D 2020 Phys. Chem. Chem. Phys. 22 16203
Google Scholar
[39] Hagan M T, Menhaj M B 1994 IEEE Trans. Neural Netw. Learn. Syst. 5 989
Google Scholar
[40] Hakalla R, Kępa R, Szajna W, Zachwieja M 2006 Eur. Phys. J. D 38 481
Google Scholar
[41] Huber K P, Herzberg G 1979 Molecular Spectra and Molecular Structure IV: Constants of Diatomic Molecules (New York: Van Nostrand Reinhold)
[42] Hase W L 1998 Classical Trajectory Simulations: Initial Conditions, A Chapter in Encyclopedia of Computational Chemistry (Vol. 1) (New York: Wiley) pp 399–402
-
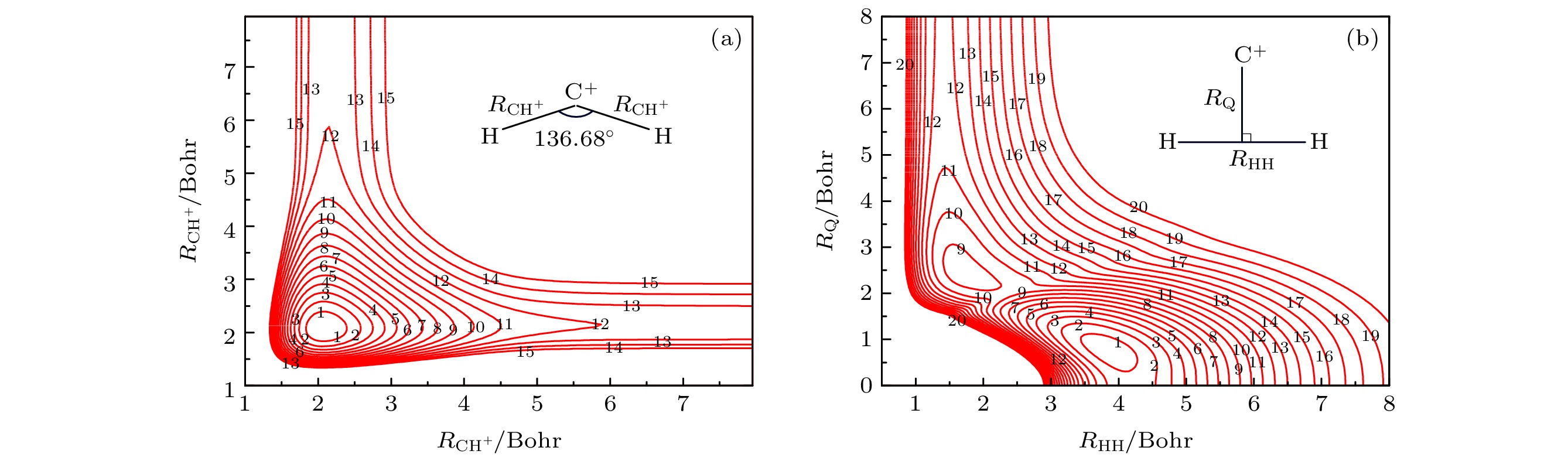
图 1 (a)键角在136.68°时, 化学键伸缩的等势线图(等势线的起点为–9.1 eV, 间隔为0.4 eV); (b) C+离子以C2v对称性接近H2分子中心的等势线图(等势线的起点为–9.1 eV, 间隔为0.37 eV)
Fig. 1. (a) Contour plot for chemical bond stretching, in which the angle is fixed at 136.68° (Contours starting at –9.1 eV and equally spaced by 0.4 eV); (b) contour plot for the C+ ion approach to the midpoint of H2 molecule in the C2v symmetry (Contours starting at –9.1 eV and equally spaced by 0.37 eV).
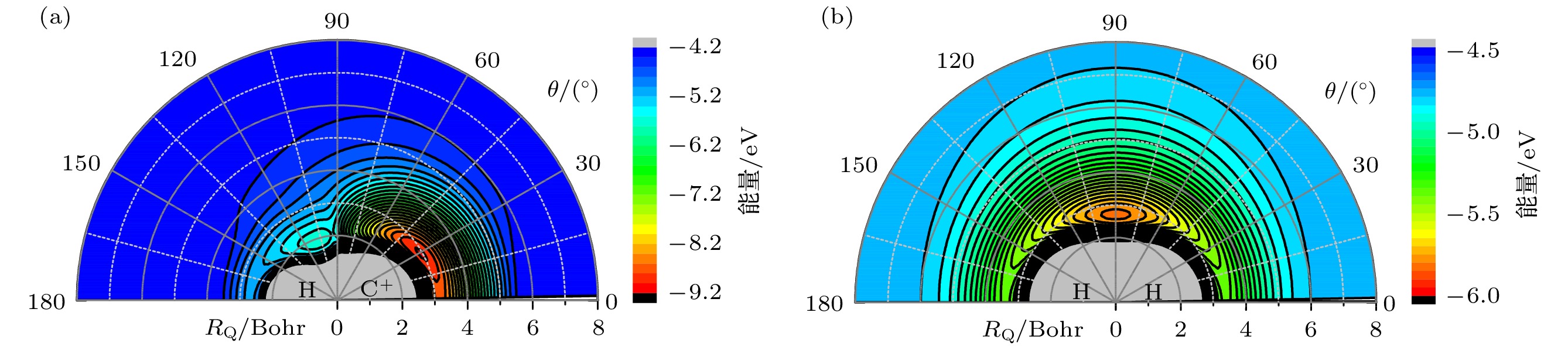
图 2 (a) 当
$ {R}_{{\mathrm{C}\mathrm{H}}^{+}}=2.136 $ Bohr时, H原子绕CH+离子运动的等势线; (b) 当$ {R}_{\mathrm{H}\mathrm{H}}=1.401 $ Bohr时, C+离子围绕H2分子运动的等势线.Fig. 2. (a) Contour plot for the H atom moves around CH+ ion at the bond distance
$ {R}_{{\mathrm{C}\mathrm{H}}^{+}}=2.136 $ Bohr; (b) contour plot for C+ ion moves around H2 molecule at its equilibrium geometry$ {R}_{\mathrm{H}\mathrm{H}}=1.401 $ Bohr.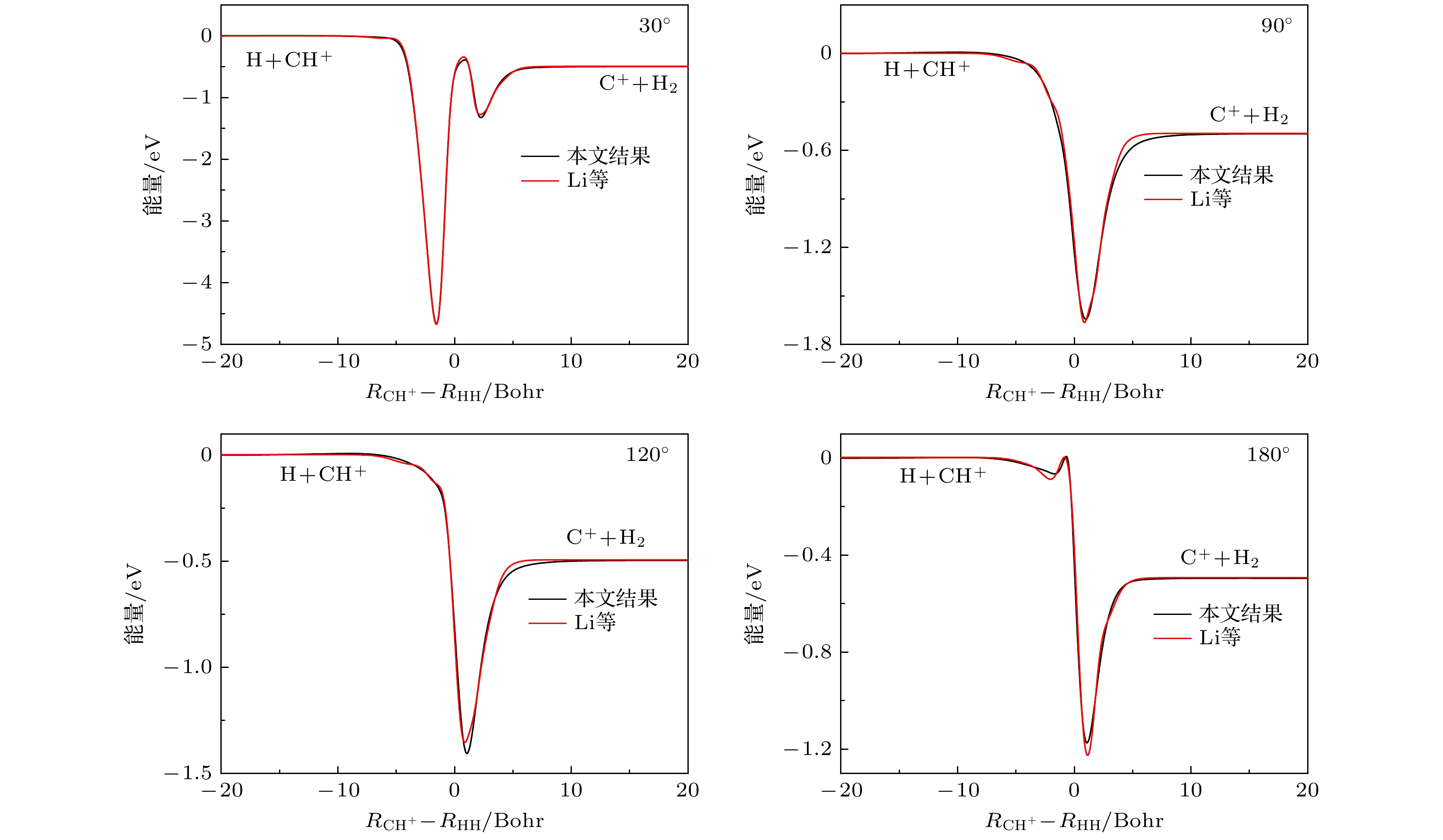


图 5 C+ + H2反应若干碰撞能下的微分截面
Fig. 5. Differential cross sections of the C+ + H2 reaction at several collision energies.
表 1 CH+和H2分子的光谱常数
Table 1. Spectroscopic constants of the CH+ and H2 molecules.
re /Bohr ωe /cm–1 ωexe /cm–1 βe /cm–1 αe /cm–1 De /eV CH+(${X}^{1}{{\Sigma } }^{+}$) 本文结果 2.136 2860.31 59.32 14.217 0.501 4.257 理论[11] 2.136 2853.03 58.52 14.201 0.489 4.252 理论[25] 2.136 2851.0 58.1 14.199 0.489 4.244 理论[26] 2.144 2849.03 66.45 14.094 0.490 理论[17] 2.135 2861.95 59.63 14.311 0.447 4.257 实验[40] 2.137 2857.56 59.32 14.178 0.495 4.26 H2(${X}^{1}{{\Sigma } }_{\mathrm{g} }^{+}$) 本文结果 1.401 4407.63 139.43 60.85 3.012 4.751 理论[17] 1.401 4404.61 126.64 60.861 2.233 4.749 理论[27] 1.401 4403.60 126.60 60.864 2.232 4.748 理论[28] 1.403 4395.22 126.12 60.735 2.221 4.748 理论[29] 1.401 4389.66 121.56 60.826 3.162 4.711 实验[41] 1.401 4401.21 121.33 60.853 3.062 4.746  下载: 导出CSV
下载: 导出CSV
-
[1] Douglas A E, Herzberg G 1941 Astrophys. J. 94 381
Google Scholar
[2] Hierl P M, Morris R A, Viggiano A A 1997 J. Chem. Phys. 106 10145
Google Scholar
[3] Plasil R, Mehner T, Dohnal P, Kotrik T, Glosik J, Gerlich D 2011 Astrophys. J. 737 60
Google Scholar
[4] Armentrout P B 2000 Int. J. Mass Spect. 200 219
Google Scholar
[5] Luca A, Borodi G, Gerlich D 2006 Photonic, Electronic and Atomic Collisions (Singapore: World Scientific)
[6] Federer W, Villinger H, Howorka F, Lindinger W, Tosi P, Bassi D, Ferguson E 1984 Phys. Rev. Lett. 52 2084
Google Scholar
[7] Stoecklin T, Halvick P 2005 Phys. Chem. Chem. Phys. 7 2446
Google Scholar
[8] Halvick P, Stoecklin T, Larrégaray P, Bonnet L 2007 Phys. Chem. Chem. Phys. 9 582
Google Scholar
[9] Zanchet A, Godard B, Bulut N, Roncero O, Halvick P, Cernicharo J 2013 Astrophys. J. 766 80
Google Scholar
[10] Warmbier R, Schneider R 2011 Phys. Chem. Chem. Phys. 13 10285
Google Scholar
[11] Li Y Q, Zhang P Y, Han K L 2015 J. Chem. Phys. 142 124302
Google Scholar
[12] Guo J, Zhang A J, Zhou Y, Liu J Y, Jia J F, Wu H S 2017 Chem. Phys. Lett. 689 121
Google Scholar
[13] Sundaram P, Manivannan V, Padamanadan R 2017 Phys. Chem. Chem. Phys. 19 20172
Google Scholar
[14] Sundaram P, Padamanadan R 2018 J. Chem. Phys. 148 164306
Google Scholar
[15] Sundaram P, Padamanadan R 2020 J. Phys. B-At. Mol. Opt. 53 105201
Google Scholar
[16] Wu H, Duan Z, Chen G 2020 Chem. Phys. Lett. 755 137783
Google Scholar
[17] Guo L, Ma H Y, Zhang L L, Song Y Z, Li Y Q 2018 RSC Adv. 8 13635
Google Scholar
[18] Herráez-Aguilar D, Jambrina P G, et al. 2014 Phys. Chem. Chem. Phys. 16 24800
Google Scholar
[19] Werfelli G, Halvick P, Honvault P, Kerkeni B, Stoecklin T 2015 J. Chem. Phys. 143 114304
Google Scholar
[20] Bovino S, Grassi T, Gianturco F A 2015 J. Phys. Chem. A 119 11973
Google Scholar
[21] Faure A, Halvick P, Stoecklin T, et al. 2017 Mon. Not. R. Astron. Soc. 469 612
Google Scholar
[22] Guo L, Yang Y F, Fan X X, Ma F C, Li Y Q 2017 Commun. Theor. Phys. 67 549
Google Scholar
[23] Gerlich D, Horning S 1992 Chem. Rev. 92 1509
Google Scholar
[24] Gerlich D, Borodi G, Luca A, Mogo C, Smith M 2011 Z. Phys. Chem. 225 475
Google Scholar
[25] Biglari Z, Shayesteh A, Maghari A 2014 Comput. Theor. Chem. 1047 22
Google Scholar
[26] Reddy R R, Nazeer A Y, Rama G K, Baba B D 2004 J. Quant. Spectrosc. Radiat. Transfer 85 105
Google Scholar
[27] Varandas A J C 1996 J. Chem. Phys. 105 3524
Google Scholar
[28] Song Y Z, Zhang Y, Zhang L L, Gao S B, Meng Q T 2015 Chin. Phys. B 24 063101
Google Scholar
[29] Yang C L, Huang Y J, Zhang X, Han K L 2003 J. Mol. Struct.: Theochem. 625 289
Google Scholar
[30] May A J, Valeev E F, Polly R, Manby F R 2005 Phys. Chem. Chem. Phys. 7 2710
Google Scholar
[31] Dunning T H 1989 J. Chem. Phys. 90 1007
Google Scholar
[32] Kendall R A, Dunning T H, Harrison R J 1992 J. Chem. Phys. 96 6796
Google Scholar
[33] Werner H J, Knowles P J 1985 J. Chem. Phys. 82 5053
Google Scholar
[34] Knowles P J, Werner H J 1985 Chem. Phys. Lett. 115 259
Google Scholar
[35] Jiang B, Li J, Guo H 2016 Int. Rev. Phys. Chem. 35 479
Google Scholar
[36] Jiang B, Guo H 2013 J. Chem. Phys. 139 054112
Google Scholar
[37] Li W T, He D, Sun Z G 2019 J. Chem. Phys. 151 185102
Google Scholar
[38] Li W T, Wang X M, Zhao H L, He D 2020 Phys. Chem. Chem. Phys. 22 16203
Google Scholar
[39] Hagan M T, Menhaj M B 1994 IEEE Trans. Neural Netw. Learn. Syst. 5 989
Google Scholar
[40] Hakalla R, Kępa R, Szajna W, Zachwieja M 2006 Eur. Phys. J. D 38 481
Google Scholar
[41] Huber K P, Herzberg G 1979 Molecular Spectra and Molecular Structure IV: Constants of Diatomic Molecules (New York: Van Nostrand Reinhold)
[42] Hase W L 1998 Classical Trajectory Simulations: Initial Conditions, A Chapter in Encyclopedia of Computational Chemistry (Vol. 1) (New York: Wiley) pp 399–402
-
[1] 赵文丽, 王永刚, 张路路, 岳大光, 孟庆田. 基于新CH2( ${\tilde {\bf{X}}{}^3}{\bf{A''}}$ )势能面的${\bf C}{\bf({}^3}{\bf{P})} + {\bf{H}_2(}{\bf X^1}\Sigma _{\bf g}^ + {\bf )} $ $ \to {\bf H({}^2}{\bf S}) + {\bf CH}{(\bf{}^2}\Pi ) $ 反应量子波包动力学. 物理学报, 2020, 69(8): 083401. doi: 10.7498/aps.69.20200132[2] 袁美玲, 李文涛. O++H2 → OH++H反应的动力学研究. 物理学报, 2019, 68(8): 083401. doi: 10.7498/aps.68.20182141 [3] 刘丽娟, 颉录有, 陈展斌, 蒋军, 董晨钟. 镁原子碰撞激发微分截面和Stokes参数的理论研究. 物理学报, 2012, 61(10): 103102. doi: 10.7498/aps.61.103102 [4] 沈光先, 汪荣凯, 令狐荣锋, 周勋, 杨向东. He-HD (HT, DT) 非对称碰撞体系振转势能面及微分散射截面的理论计算. 物理学报, 2012, 61(21): 213101. doi: 10.7498/aps.61.213101 [5] 李勇军, 冯灏, 孙卫国, 曾阳阳, 王小炼, 李会东, 樊群超. 基于严格交换势的低能电子与H2分子碰撞振动激发散射截面的研究. 物理学报, 2011, 60(4): 043401. doi: 10.7498/aps.60.043401 [6] 李劲, 令狐荣锋, 司冠杰, 杨向东. 低能He原子与Li2分子碰撞散射截面理论计算. 物理学报, 2010, 59(8): 5424-5428. doi: 10.7498/aps.59.5424 [7] 王悦, 董德智, 李伟艳, 凤尔银, 崔执凤. He-Na2体系低温下的冷碰撞研究. 物理学报, 2009, 58(10): 6913-6919. doi: 10.7498/aps.58.6913 [8] 王斌, 冯灏, 孙卫国, 曾阳阳, 戴伟. 低能电子与氢分子碰撞的振动激发积分散射截面的研究. 物理学报, 2009, 58(10): 6932-6937. doi: 10.7498/aps.58.6932 [9] 余春日, 汪荣凯, 张杰, 杨向东. He同位素原子与HBr分子碰撞的微分截面. 物理学报, 2009, 58(1): 229-233. doi: 10.7498/aps.58.229 [10] 汪荣凯, 沈光先, 杨向东. He-BH碰撞体系微分截面的理论计算. 物理学报, 2009, 58(8): 5335-5341. doi: 10.7498/aps.58.5335 [11] 潘 宇, 王凯俊, 方祯云, 汪先友, 彭庆军. 精确计算n-n重正化链图传播下n+n→2π0反应截面. 物理学报, 2008, 57(8): 4817-4825. doi: 10.7498/aps.57.4817 [12] 王 平, 李芳昱, 何晓宇. 电磁场中光子-轴子的转化微分截面. 物理学报, 2008, 57(9): 5442-5447. doi: 10.7498/aps.57.5442 [13] 汪荣凯, 沈光先, 宋晓书, 令狐荣锋, 杨向东. He同位素对He-NO碰撞体系微分截面的影响. 物理学报, 2008, 57(7): 4138-4142. doi: 10.7498/aps.57.4138 [14] 余春日, 凤尔银, 程新路, 杨向东. He-HI复合物势能面及微分散射截面的理论研究. 物理学报, 2007, 56(8): 4441-4447. doi: 10.7498/aps.56.4441 [15] 施德恒, 孙金锋, 朱遵略, 杨向东, 刘玉芳, 马 恒. 中、高能电子被SO2分子散射的微分截面、动量转移截面及弹性积分截面. 物理学报, 2007, 56(8): 4435-4440. doi: 10.7498/aps.56.4435 [16] 汪荣凯, 令狐荣锋, 杨向东. He-NO碰撞体系微分截面的理论计算. 物理学报, 2007, 56(4): 2067-2072. doi: 10.7498/aps.56.2067 [17] 韩慧仙, 彭 谦, 文振翼, 王育彬. S2O分子的局域势能面和振动光谱的解析. 物理学报, 2005, 54(1): 78-84. doi: 10.7498/aps.54.78 [18] 施德恒, 孙金锋, 朱遵略, 刘玉芳, 杨向东. 中高能电子被O2及CF4分子散射的微分截面、弹性积分截面及动量转移截面. 物理学报, 2005, 54(8): 3548-3553. doi: 10.7498/aps.54.3548 [19] 王晓艳, 丁世良. 用李代数方法构造四原子分子的势能面. 物理学报, 2004, 53(2): 423-426. doi: 10.7498/aps.53.423 [20] 孙桂华, 杨向东. H+H2反应截面的全量子力学研究. 物理学报, 2002, 51(3): 506-511. doi: 10.7498/aps.51.506


 下载:
下载:









计量
- 文章访问数: 7643
- PDF下载量: 92
- 被引次数: 0
