










-
In the present work, the long-range interaction potential part of potential energy surface (PES) of OH2+ system is revised and the new resulting PES apparently is more reasonable than the old one in the long-range part. Based on the new PES, the dynamics calculations of O+ +H2→ OH+ + H reaction are carried out at a state-to-state level of theory by using time-dependent quantum wave packet method with second order split operator in a collision energy range from 0.01 to 1.0 eV. The dynamic properties such as reaction probability, ro-vibrational resolved statereaction probability, integral cross section, differential cross section, and state specific rate constant are calculated and compared with available theoretical and experimental results. The results of ro-vibrational resolved state reaction probability reflect some dynamic properties such as resonances which is attributed to the deep well located on the reaction path. The vibrational resolved state reaction probability indicates that the excitation efficiency of the OH+ product is relatively low. The results of integral cross sections indicate that the present results are in better agreement with the experimental values than with previous theoretical calculations, especially in the low collision energy region. However, the state specific rate constant results underestimate the experimental values. The comparison betweenour calculations and the experimental results indicates that the contribution of the rotational excitation of H2 molecule should be included in the calculations. However, only the initial state v = 0, j = 0 is calculated in the present work. We suppose that the deviation of the present results from the experimental data is due to the fact that the rotational excitation of reactant isnot included in the present calculation. The differential cross section signals indicate that the complex-forming reaction mechanism isdominated in the case of low collision energy, but it transforms into abstract reaction mechanism as the collision energy further increases.
[1] Duley W W, Williams D A 1984 Interstellar Chemistry (New York: Academic Press) p251
[2] Armentrout P 2000 Int. J. Mass Spectrom. Ion Process. 200 21933
[3] Jambrina P G, Alvarino J M, Gerlich D, Hankel M, Herrero V J, Saez-Rabanos V, Aoiz F J 2012 Phys. Chem. Chem. Phys. 14 3346
 Google Scholar
Google Scholar
[4] Wang B B, Han Y C, Gao W, Cong S L 2017 Phys. Chem. Chem. Phys. 19 22926
Google Scholar
[5] Fehsenfeld F C, Schmeltekopf A L, Ferguson E E 1967 J. Chem. Phys. 46 2802
Google Scholar
[6] Kim J K, Theard L P, Huntress W T 1975 J. Chem. Phys. 62 45
Google Scholar
[7] Federer W, Villinger H, Howorka F, Lindinger W, Tosi P, Bassi D, Ferguson E 1984 Phys. Rev. Lett. 52 2084
Google Scholar
[8] Smith D, Adams N G, Miller T M 1978 J. Chem. Phys. 69 308
Google Scholar
[9] Burley J, Ervin K M, Armentrout P 1987 Int. J. Mass Spectrom. Ion Process 80 153
Google Scholar
[10] Sunderlin L, Armentrout P 1990 Chem. Phys. Lett. 167 188
Google Scholar
[11] Flesch G D, Ng C Y 1991 J. Chem. Phys. 94 2372
Google Scholar
[12] Li X, Huang Y L, Flesch G D, Ng C Y 1997 J. Chem. Phys. 106 564
Google Scholar
[13] Gillen K T, Mahan B H, Winn J S 1973 J. Chem. Phys. 58 5373
Google Scholar
[14] Gillen K T, Mahan B H, Winn J S 1973 J. Chem. Phys. 59 6380
Google Scholar
[15] Ng C Y 2002 J. Phys. Chem. A 106 5953
Google Scholar
[16] Martínez R, Millán J, González M 2004 J. Chem. Phys. 120 4705
Google Scholar
[17] Martínez R, Sierra J D, González M 2005 J. Chem. Phys. 123 174312
Google Scholar
[18] Martínez R, Lucas J M, Giménez X, Aguilar A, González M 2006 J. Chem. Phys. 124 144301
Google Scholar
[19] Martínez R, Sierra J D, Gray S K, González M 2006 J. Chem. Phys. 125 164305
Google Scholar
[20] Kłos J, Bulut N, Akpinar S 2012 Chem. Phys. Lett. 532 22
Google Scholar
[21] Xu W, Li W, Lv S, Zhai H, Duan Z, Zhang P 2012 J. Phys. Chem. A 116 10882
Google Scholar
[22] Gómez-Carrasco S, Godard B, Lique F, Bulut N, Kłos J, Roncero O, Aguado A, Aoiz F J, Castillo J F, Goicoechea J R 2014 Astrophys. J. 794 33
Google Scholar
[23] Bulut N, Castillo J F, Jambrina P G, Kłos J, Roncero O, Aoiz F J, Bañares L 2015 J. Phys. Chem. A 119 11951
Google Scholar
[24] Li W T, Yuan J C, Yuan M L, Zhang Y, Yao M H, Sun Z G 2018 Phys. Chem. Chem. Phys. 20 1039
Google Scholar
[25] 段志欣, 邱明辉, 姚翠霞 2014 物理学报 63 063402
Google Scholar
Duan Z X, Qiu M H, Yao C X 2014 Acta Phys. Sin. 63 063402
Google Scholar
[26] 李文涛, 于文涛, 姚明海 2018 物理学报 67 103401
Google Scholar
Li W T, Yu W T, Yao M H 2018 Acta Phys. Sin. 67 103401
Google Scholar
[27] 张静, 魏巍, 高守宝, 孟庆田 2015 物理学报 64 063101
Google Scholar
Zhang J, Wei W, Gao S B, Meng Q T 2015 Acta Phys. Sin. 64 063101
Google Scholar
[28] Zhao B, Sun Z G, Guo H 2016 J. Chem. Phys. 144 214303
Google Scholar
[29] Zhao B, Sun Z G, Guo H 2016 J. Chem. Phys. 144 064104
Google Scholar
[30] Li W T, Chen M D, Sun Z G 2015 Chin. J. Chem. Phys. 28 415
Google Scholar
[31] Fleck J A, Morris J R, Feit M D 1976 Appl. Phys. 10 129
-

图 1 新旧两个势能面的长程相互作用势比较
Fig. 1. Comparison of the long range interaction potential for the old and new potential energy surfaces

图 2 90°, 120°, 150°和180°的最小能量路径
Fig. 2. The minimum energy paths at 90°, 120°, 150°, and 180° angles
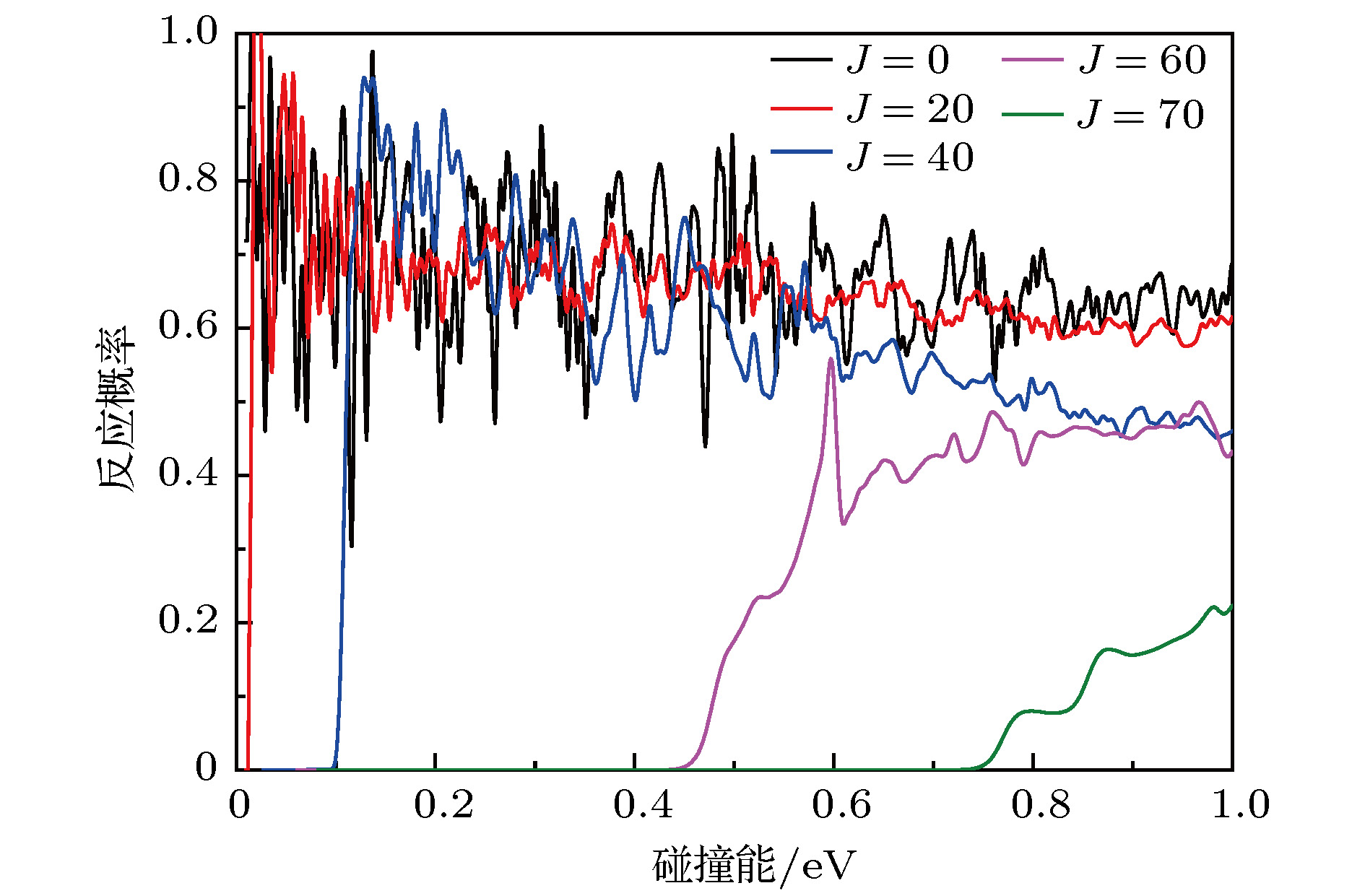
图 3 对于几个选定的总角动量J在能量范围0—1.0 eV的反应概率
Fig. 3. The total reaction probabilities for several selected total angular momentum J in the collision energy from 0 to 1.0 eV
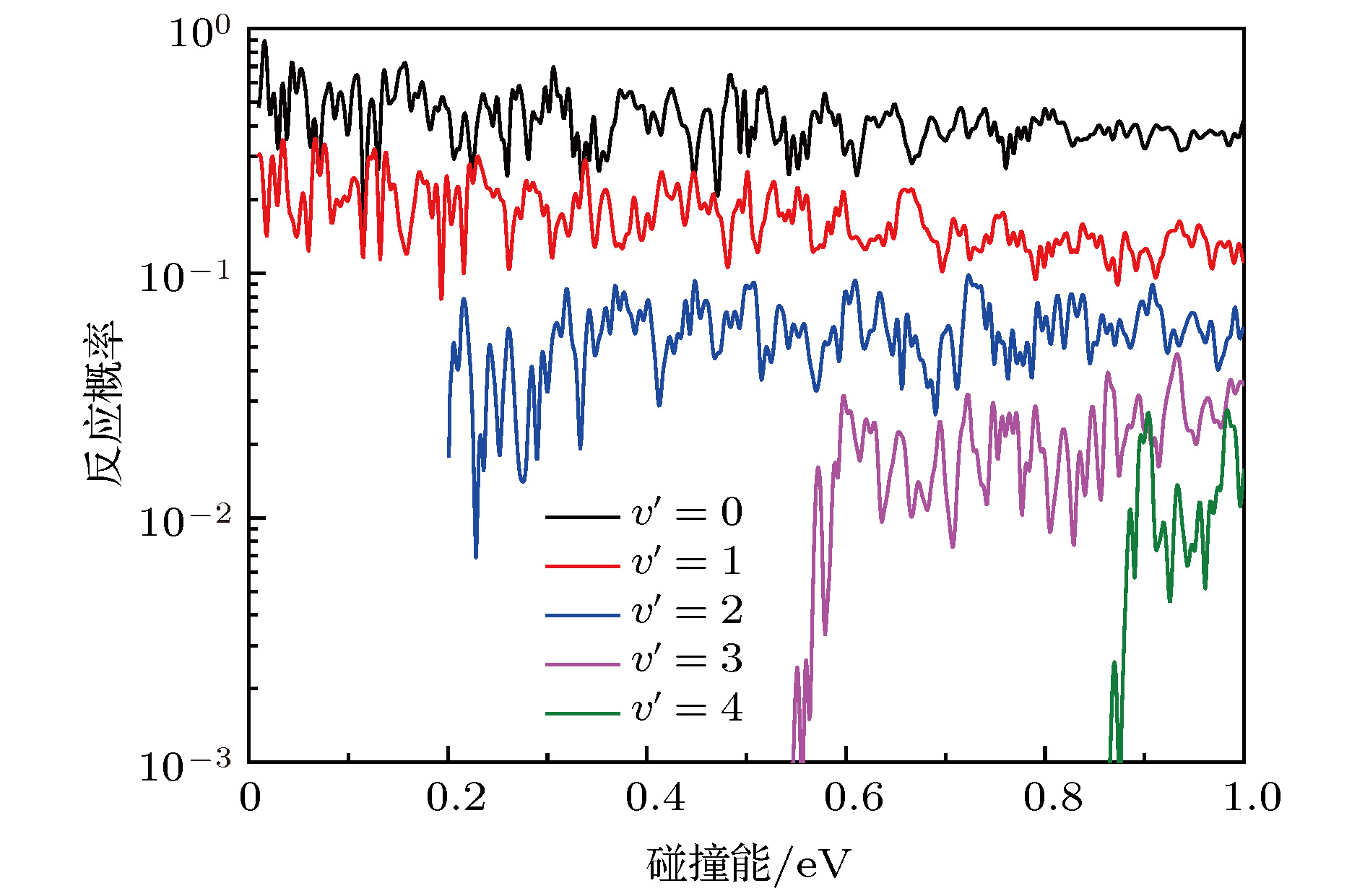
图 4 总角动量J = 0反应概率的振动分辨
Fig. 4. The vibrational resolved reaction probabilities of the total angular momentum J = 0
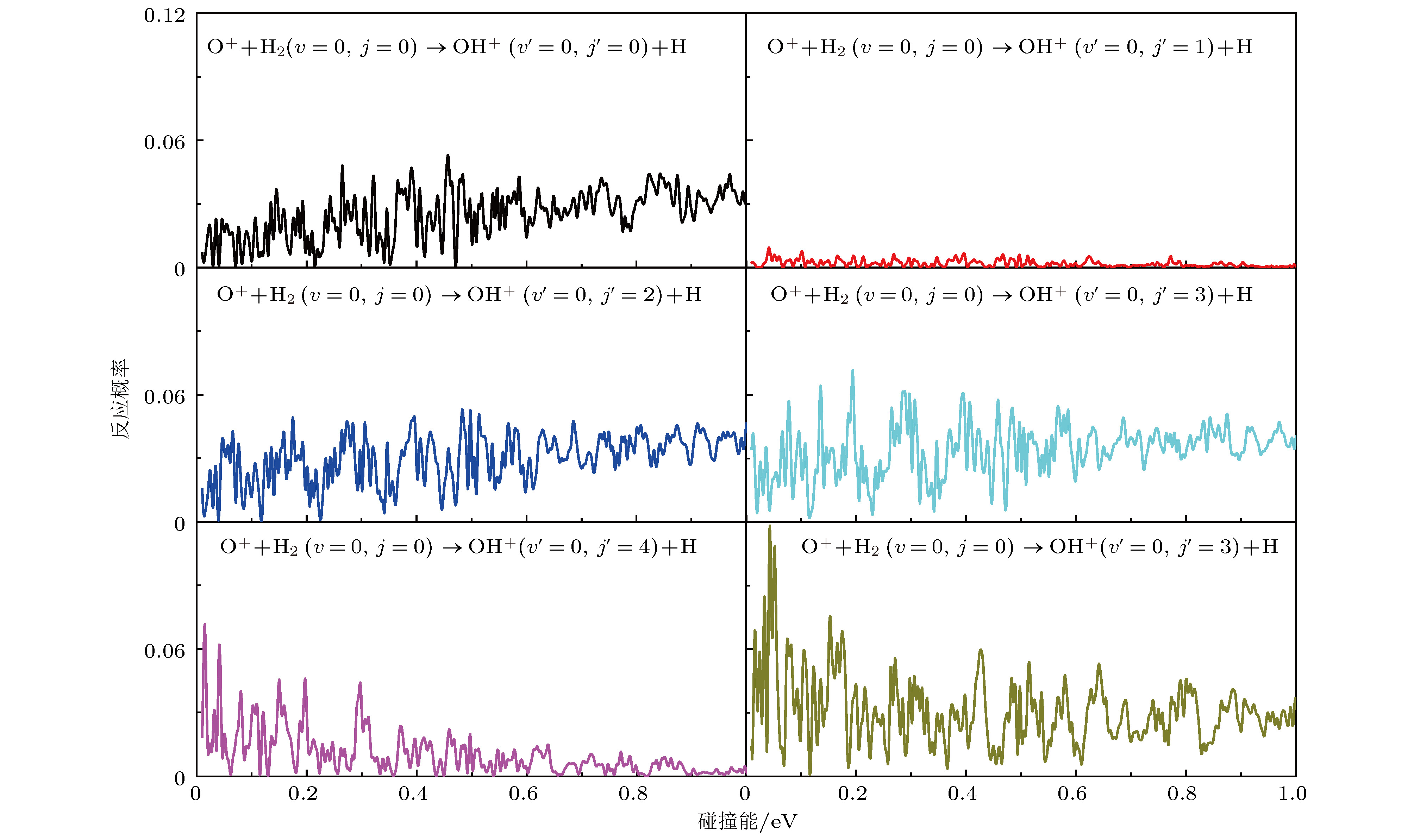
图 5 O+ + H2(v = 0, j = 0) → OH+ (v' = 0, j' ) + H反应在总角动量J = 0时转动分辨的反应概率
Fig. 5. Rotationally resolved reaction probabilities for the O+ + H2 (v = 0, j = 0) → OH+(v' = 0, j' ) + H reaction at total angular momentum J = 0
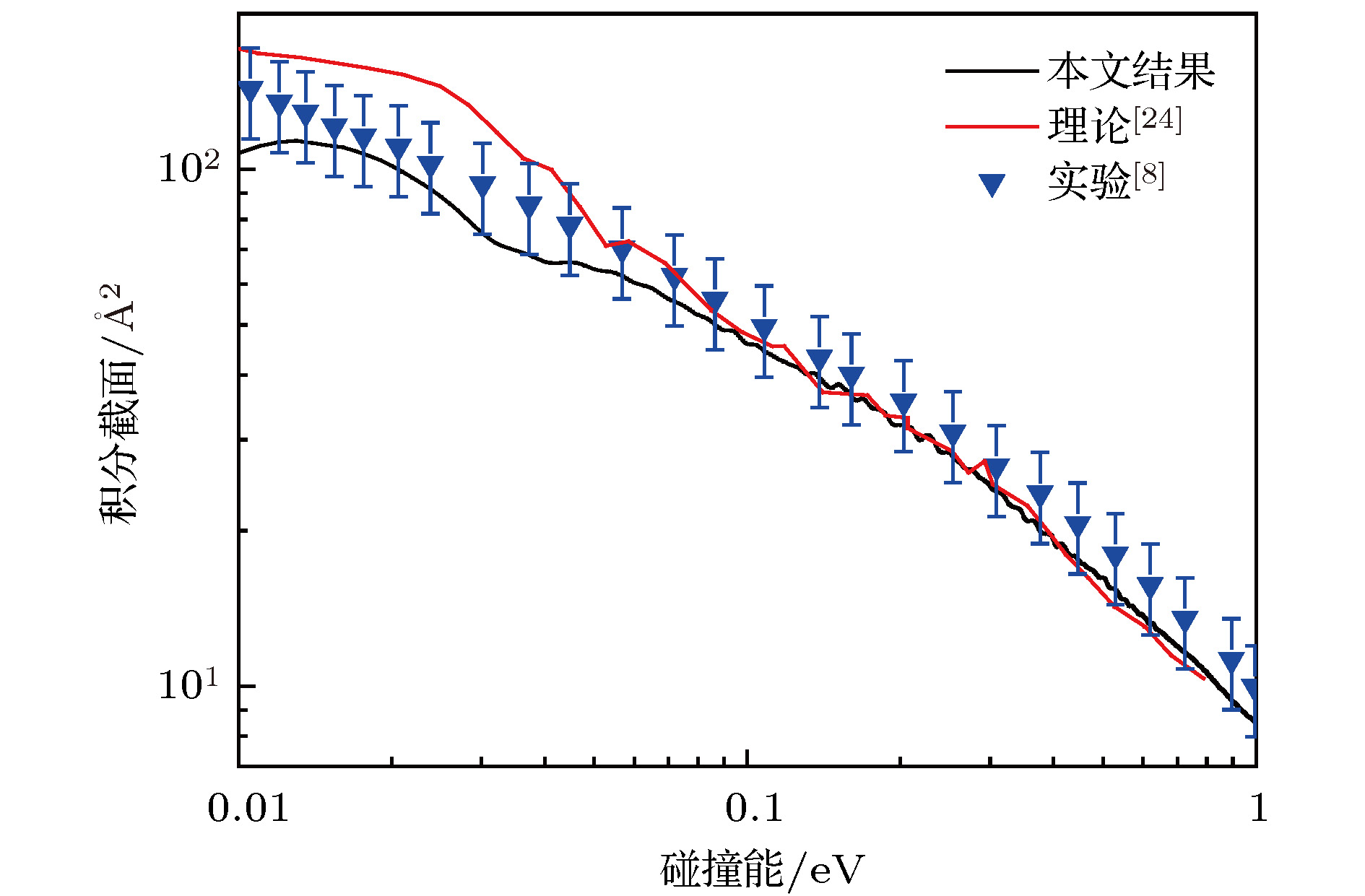
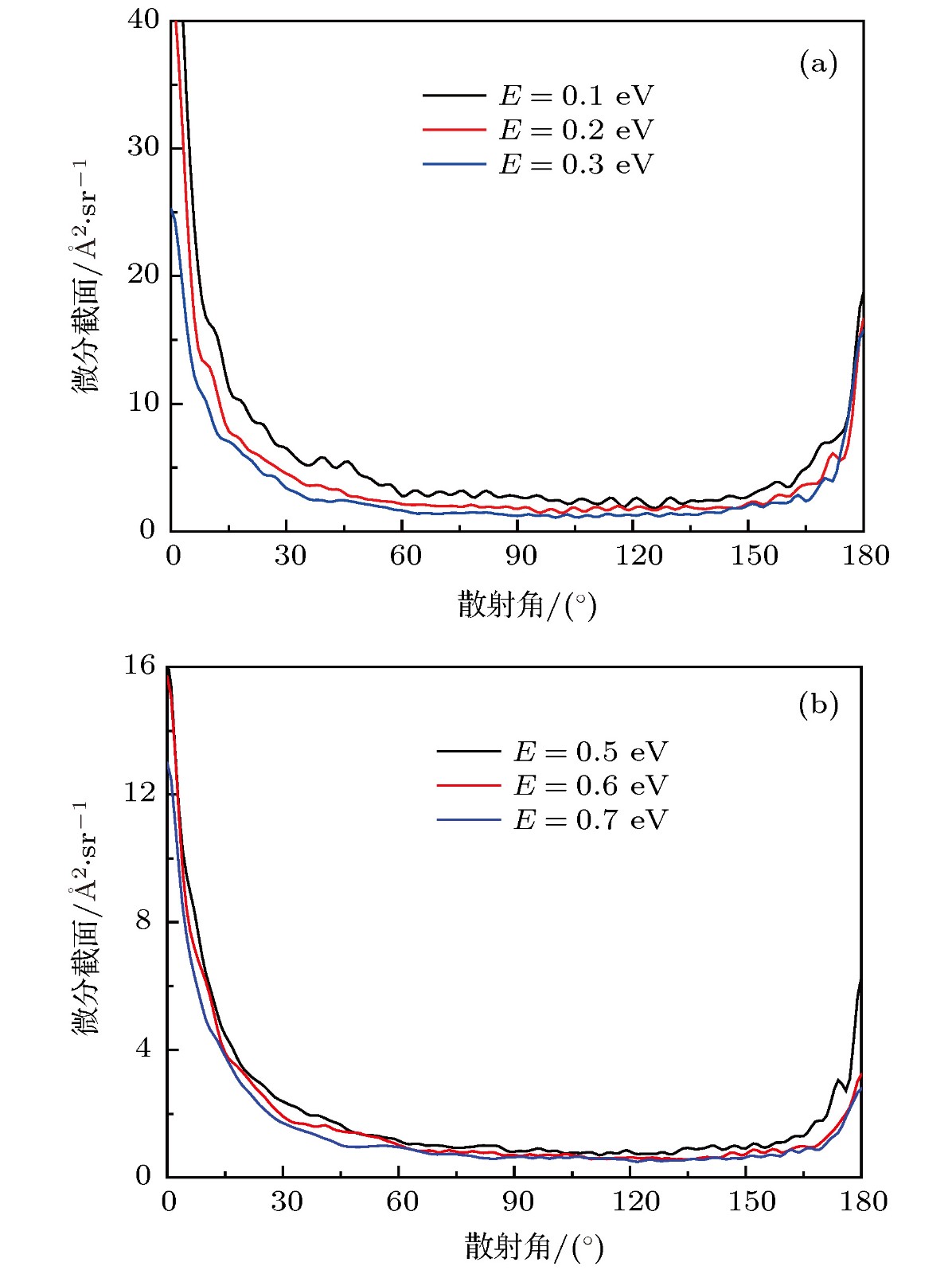
图 7 O+ + H2反应若干能量点的微分截面 (a)低能部分; (b) 高能部分
Fig. 7. The differential cross sections of O+ + H2 reaction for several collision energies: (a) The low energy; (b) the high energy part

表 1 计算中使用的参数(除了特殊声明, 均采用原子单位a.u.)
Table 1. Parameters used in the calculation (The atomic unit is used in the calculation unless otherwise stated)
格点范围和大小 $R \in \left[ {0.1,16} \right],{N_R} = 279,N_R^{{\rm{int}}} = 159$ $r \in \left[ {0.1,15} \right],{N_r} = 279,N_r^{{\rm{asy}}} = 159$ 初始波包 Rc = 11.0, ${k_0} = \sqrt {2{E_0}{\mu_R}} $ ${\varDelta _R} = 0.2 {\rm{exp}}\left[ { - \dfrac{{{{\left( {R - {R_{\rm{c}}}} \right)}^2}}}{{2\varDelta _R^2}}} \right]{\rm{cos}}\left( {{k_0}R} \right)$;
其中E0 = 0.5 eV总传播时间 30000 最大总角
动量J70  下载: 导出CSV
下载: 导出CSV
-
[1] Duley W W, Williams D A 1984 Interstellar Chemistry (New York: Academic Press) p251
[2] Armentrout P 2000 Int. J. Mass Spectrom. Ion Process. 200 21933
[3] Jambrina P G, Alvarino J M, Gerlich D, Hankel M, Herrero V J, Saez-Rabanos V, Aoiz F J 2012 Phys. Chem. Chem. Phys. 14 3346
Google Scholar
[4] Wang B B, Han Y C, Gao W, Cong S L 2017 Phys. Chem. Chem. Phys. 19 22926
Google Scholar
[5] Fehsenfeld F C, Schmeltekopf A L, Ferguson E E 1967 J. Chem. Phys. 46 2802
Google Scholar
[6] Kim J K, Theard L P, Huntress W T 1975 J. Chem. Phys. 62 45
Google Scholar
[7] Federer W, Villinger H, Howorka F, Lindinger W, Tosi P, Bassi D, Ferguson E 1984 Phys. Rev. Lett. 52 2084
Google Scholar
[8] Smith D, Adams N G, Miller T M 1978 J. Chem. Phys. 69 308
Google Scholar
[9] Burley J, Ervin K M, Armentrout P 1987 Int. J. Mass Spectrom. Ion Process 80 153
Google Scholar
[10] Sunderlin L, Armentrout P 1990 Chem. Phys. Lett. 167 188
Google Scholar
[11] Flesch G D, Ng C Y 1991 J. Chem. Phys. 94 2372
Google Scholar
[12] Li X, Huang Y L, Flesch G D, Ng C Y 1997 J. Chem. Phys. 106 564
Google Scholar
[13] Gillen K T, Mahan B H, Winn J S 1973 J. Chem. Phys. 58 5373
Google Scholar
[14] Gillen K T, Mahan B H, Winn J S 1973 J. Chem. Phys. 59 6380
Google Scholar
[15] Ng C Y 2002 J. Phys. Chem. A 106 5953
Google Scholar
[16] Martínez R, Millán J, González M 2004 J. Chem. Phys. 120 4705
Google Scholar
[17] Martínez R, Sierra J D, González M 2005 J. Chem. Phys. 123 174312
Google Scholar
[18] Martínez R, Lucas J M, Giménez X, Aguilar A, González M 2006 J. Chem. Phys. 124 144301
Google Scholar
[19] Martínez R, Sierra J D, Gray S K, González M 2006 J. Chem. Phys. 125 164305
Google Scholar
[20] Kłos J, Bulut N, Akpinar S 2012 Chem. Phys. Lett. 532 22
Google Scholar
[21] Xu W, Li W, Lv S, Zhai H, Duan Z, Zhang P 2012 J. Phys. Chem. A 116 10882
Google Scholar
[22] Gómez-Carrasco S, Godard B, Lique F, Bulut N, Kłos J, Roncero O, Aguado A, Aoiz F J, Castillo J F, Goicoechea J R 2014 Astrophys. J. 794 33
Google Scholar
[23] Bulut N, Castillo J F, Jambrina P G, Kłos J, Roncero O, Aoiz F J, Bañares L 2015 J. Phys. Chem. A 119 11951
Google Scholar
[24] Li W T, Yuan J C, Yuan M L, Zhang Y, Yao M H, Sun Z G 2018 Phys. Chem. Chem. Phys. 20 1039
Google Scholar
[25] 段志欣, 邱明辉, 姚翠霞 2014 物理学报 63 063402
Google Scholar
Duan Z X, Qiu M H, Yao C X 2014 Acta Phys. Sin. 63 063402
Google Scholar
[26] 李文涛, 于文涛, 姚明海 2018 物理学报 67 103401
Google Scholar
Li W T, Yu W T, Yao M H 2018 Acta Phys. Sin. 67 103401
Google Scholar
[27] 张静, 魏巍, 高守宝, 孟庆田 2015 物理学报 64 063101
Google Scholar
Zhang J, Wei W, Gao S B, Meng Q T 2015 Acta Phys. Sin. 64 063101
Google Scholar
[28] Zhao B, Sun Z G, Guo H 2016 J. Chem. Phys. 144 214303
Google Scholar
[29] Zhao B, Sun Z G, Guo H 2016 J. Chem. Phys. 144 064104
Google Scholar
[30] Li W T, Chen M D, Sun Z G 2015 Chin. J. Chem. Phys. 28 415
Google Scholar
[31] Fleck J A, Morris J R, Feit M D 1976 Appl. Phys. 10 129
-
[1] 赵文丽, 宋玉志, 马超, 高峰, 孟庆田. 基于一个新SiH2(11A′)势能面的H+SiH反应动力学研究. 物理学报, 2024, 73(20): 203401. doi: 10.7498/aps.73.20240859 [2] 赵文丽, 孙丰伟, 张红, 王永刚, 高峰, 孟庆田. $ {\text{D}} + {\text{Si}}{{\text{D}}^ + } \to {{\text{D}}_2} + {\text{S}}{{\text{i}}^ + } $ 反应量子波包动力学研究. 物理学报, 2022, 71(22): 228201. doi: 10.7498/aps.71.20221155[3] 李文涛, 袁美玲, 王杰敏. C++H2反应的动力学研究: 基于一个新构建的势能面. 物理学报, 2022, 71(9): 093402. doi: 10.7498/aps.71.20212241 [4] 袁方园, 朱子亮. D + DBr反应的态-态动力学研究. 物理学报, 2020, 69(11): 113401. doi: 10.7498/aps.69.20200321 [5] 李文涛, 于文涛, 姚明海. 采用量子含时波包方法研究H/D+Li2LiH/LiD+Li反应. 物理学报, 2018, 67(10): 103401. doi: 10.7498/aps.67.20180324 [6] 张静, 魏巍, 高守宝, 孟庆田. H+Li2: 一个典型的释能反应体系及其含时动力学研究. 物理学报, 2015, 64(6): 063101. doi: 10.7498/aps.64.063101 [7] 刘丽娟, 颉录有, 陈展斌, 蒋军, 董晨钟. 镁原子碰撞激发微分截面和Stokes参数的理论研究. 物理学报, 2012, 61(10): 103102. doi: 10.7498/aps.61.103102 [8] 沈光先, 汪荣凯, 令狐荣锋, 周勋, 杨向东. He-HD (HT, DT) 非对称碰撞体系振转势能面及微分散射截面的理论计算. 物理学报, 2012, 61(21): 213101. doi: 10.7498/aps.61.213101 [9] 李勇军, 冯灏, 孙卫国, 曾阳阳, 王小炼, 李会东, 樊群超. 基于严格交换势的低能电子与H2分子碰撞振动激发散射截面的研究. 物理学报, 2011, 60(4): 043401. doi: 10.7498/aps.60.043401 [10] 李劲, 令狐荣锋, 司冠杰, 杨向东. 低能He原子与Li2分子碰撞散射截面理论计算. 物理学报, 2010, 59(8): 5424-5428. doi: 10.7498/aps.59.5424 [11] 王斌, 冯灏, 孙卫国, 曾阳阳, 戴伟. 低能电子与氢分子碰撞的振动激发积分散射截面的研究. 物理学报, 2009, 58(10): 6932-6937. doi: 10.7498/aps.58.6932 [12] 余春日, 汪荣凯, 张杰, 杨向东. He同位素原子与HBr分子碰撞的微分截面. 物理学报, 2009, 58(1): 229-233. doi: 10.7498/aps.58.229 [13] 汪荣凯, 沈光先, 杨向东. He-BH碰撞体系微分截面的理论计算. 物理学报, 2009, 58(8): 5335-5341. doi: 10.7498/aps.58.5335 [14] 王 平, 李芳昱, 何晓宇. 电磁场中光子-轴子的转化微分截面. 物理学报, 2008, 57(9): 5442-5447. doi: 10.7498/aps.57.5442 [15] 潘 宇, 王凯俊, 方祯云, 汪先友, 彭庆军. 精确计算n-n重正化链图传播下n+n→2π0反应截面. 物理学报, 2008, 57(8): 4817-4825. doi: 10.7498/aps.57.4817 [16] 汪荣凯, 沈光先, 宋晓书, 令狐荣锋, 杨向东. He同位素对He-NO碰撞体系微分截面的影响. 物理学报, 2008, 57(7): 4138-4142. doi: 10.7498/aps.57.4138 [17] 施德恒, 孙金锋, 朱遵略, 杨向东, 刘玉芳, 马 恒. 中、高能电子被SO2分子散射的微分截面、动量转移截面及弹性积分截面. 物理学报, 2007, 56(8): 4435-4440. doi: 10.7498/aps.56.4435 [18] 汪荣凯, 令狐荣锋, 杨向东. He-NO碰撞体系微分截面的理论计算. 物理学报, 2007, 56(4): 2067-2072. doi: 10.7498/aps.56.2067 [19] 施德恒, 孙金锋, 朱遵略, 刘玉芳, 杨向东. 中高能电子被O2及CF4分子散射的微分截面、弹性积分截面及动量转移截面. 物理学报, 2005, 54(8): 3548-3553. doi: 10.7498/aps.54.3548 [20] 白丽华, 张庆刚, 刘新国. D+CD4→CD3+D2反应的四维量子散射计算. 物理学报, 2003, 52(11): 2774-2780. doi: 10.7498/aps.52.2774

 下载:
下载:

计量
- 文章访问数: 11021
- PDF下载量: 62
- 被引次数: 0
