










-
Electrolyte not only plays the role of conducting ions in lithium ion battery, but also the thin layer electrolyte formed on the electrode surface determines the stability of electrode/electrolyte interface to a large extent, thus affecting the cycling stability, rate performance and safety of the battery. The successful commercialization and widespread application of lithium ion battery is closely related to the solid electrolyte interface film formed by the decomposition of electrolyte on the electrode surface. In this paper, the electrochemical stability and decomposition mechanism of the interface electrolyte are briefly reviewed, aiming to draw more scientists' attention to the electrolyte and its interfacial properties.
-
Keywords:
- lithium ion battery /
- electrode/electrolyte interface /
- electrochemical stabiity /
- solid electrolyte interphase film /
- mechanism study
[1] Nitta N, Wu F, Lee J T, Yushin G 2015 Mater. Today 18 252
 Google Scholar
Google Scholar
[2] Xu K 2004 Chem. Rev. 104 4303
Google Scholar
[3] Xu K 2014 Chem. Rev. 114 11503
Google Scholar
[4] Winter M, Barnett B, Xu K 2018 Chem. Rev. 118 11433
Google Scholar
[5] Zhang X Q, Chen X, Hou L P, Li B Q, Cheng X B, Huang J Q, Zhang Q 2019 ACS Energy Lett. 4 411
Google Scholar
[6] Chen X, Zhang Q 2020 Acc. Chem. Res. 53 1992
Google Scholar
[7] Winter M 2009 Z. Phys. Chem. 223 1395
Google Scholar
[8] Borodin O, Smith G D 2009 J. Phys. Chem. B 113 1763
Google Scholar
[9] Xing L D, Li W S, Wang C Y, Xu M Q, Tan C L, Yi J 2009 J. Phys. Chem. B 113 16596
[10] Xing L D, Wang C Y, Xu M Q, Li W S, Cai Z P 2009 J. Power Sources 189 689
[11] Xing L D, Wang C Y, Li W S, Xu M Q, Meng X L, Zhao S F 2009 J. Phys. Chem. B 113 5181
[12] Wang Q, Evans N, Zakeeruddin S M, Exnar I, Gra¨tzel M 2007 J. Am. Chem. Soc. 129 3163
[13] Xing L D, Borodin O, Smith G, Li W S 2011 J. Phys. Chem. A 115 13896
Google Scholar
[14] Borodin O, Jow T R 2011 ECS Trans. 33 77
[15] Vatamanu J, Borodin O, Smith G D 2012 J. Phys. Chem. C 116 1114
Google Scholar
[16] Xing L D, Vatamanu J, Bedrov D, Borodin O, Smith G D 2012 J. Phys. Chem. C 116 23871
Google Scholar
[17] Watanabe Y, Kinoshita S I, Wada S, Hoshino K, Morimoto H, Tobishima S I 2008 J. Power Sources 179 770
Google Scholar
[18] Fan X, Ji X, Chen L, Chen J, Deng T, Han F, Yue J, Piao N, Wang R, Zhou X, Xiao X, Chen L, Wang C 2019 Nat. Energy 4 882
Google Scholar
[19] Xing L D, Borodin O 2012 Phys. Chem. Chem. Phys. 14 12838
Google Scholar
[20] Abu-Lebdeh Y, Davidson I 2009 J. Electrochem. Soc. 156 A60
Google Scholar
[21] Kim Y S, Kim T H, Lee H, Song H K 2011 Energy Environ. Sci. 4 4038
Google Scholar
[22] Zhi H Z, Xing L D, Zheng X W, Xu K, Li W S 2017 J. Phys. Chem. Lett. 8 6048
Google Scholar
[23] Wang Y T, Xing L D, Li W S, Bedrov D 2013 J. Phys. Chem. Lett. 4 3992
Google Scholar
[24] Zhuang G V, Yang H, Blizanac B, Ross P N 2015 Electrochem. Solid-State Lett. 8 A441
[25] Tasaki K, Goldberg A, Winter M 2011 Electrochim. Acta 56 10424
Google Scholar
[26] Xing L D, Zheng X W, Schroeder M, Alvarado J, Cresce A W, Xu K 2018 Acc. Chem. Res. 51 282
Google Scholar
[27] Wang C, Xing L D, Vatamanu J, Chen Z, Lan G Y, Li W S, Xu K 2019 Nat. Commun. 10 3423
Google Scholar
[28] Jarry A, Gottis S, Yu Y S, Roque R J, Kim C, Cabana J, Kerr J, Kostecki R 2015 J. Am. Chem. Soc. 137 3533
Google Scholar
[29] Zhan C, Lu J, Kropf A J, Wu T P, Jansen A N, Sun Y K, Qiu X P, Amine K 2013 Nat. Commun. 4 2437
Google Scholar
[30] Wang K, Xing L D, Xu K, Zhou H B, Li W S 2019 ACS Appl. Mater. Interfaces 11 31490
Google Scholar
[31] Zhu Y M, Rong H B, Mai S W, Luo X Y, Li X P, Li W S 2015 J. Power Sources 299 485
Google Scholar
[32] Zheng Q F, Xing L D, Yang X R, Li X F, Ye C C, Wang K, Huang Q M, Li W S 2018 ACS Appl. Mater. Interfaces 10 16843
Google Scholar
[33] Chen J W, Xing L D, Yang X R, Liu X, Li T J, Li W S 2018 Electrochim. Acta 290 568
Google Scholar
-

图 1 锂离子电池的工作电压窗口与SEI/CEI膜的关系
Fig. 1. Relationship between electrochemical window and SEI/CEI film of lithium-ion batteries.
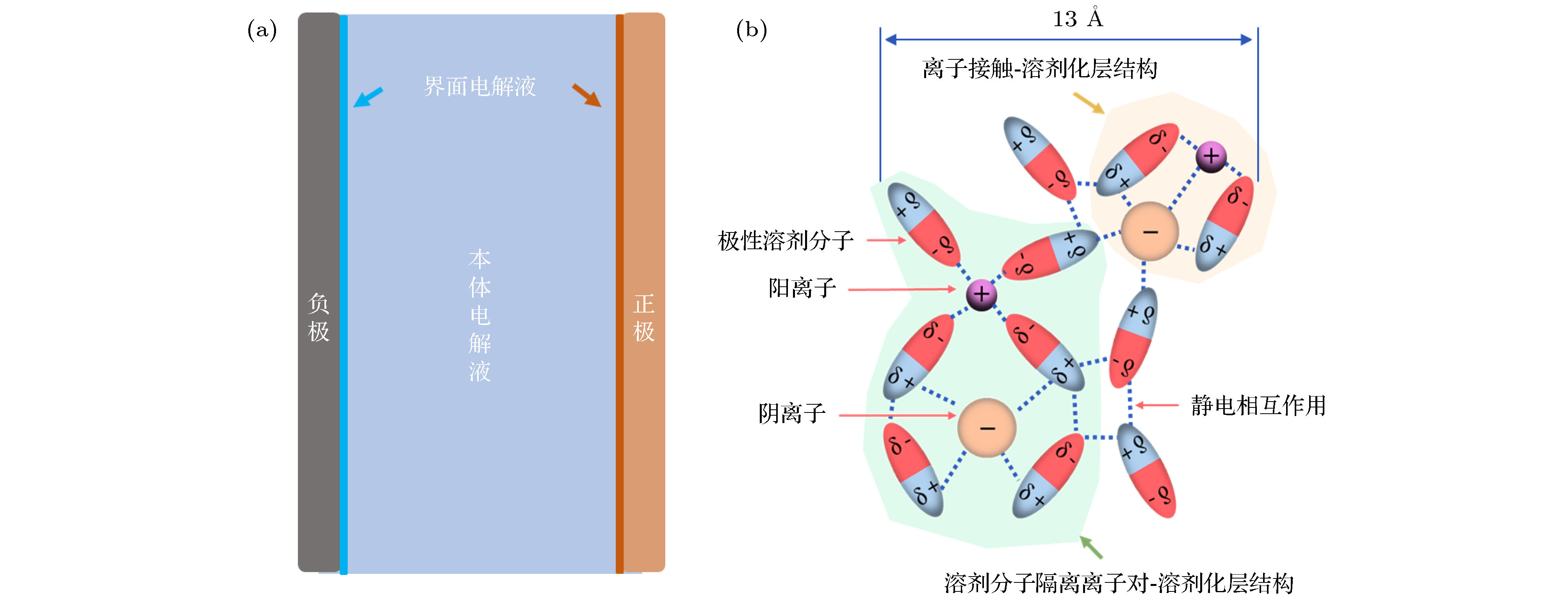
图 2 (a) 电池中界面电解液与本体电解液的差异; (b) 界面电解液的主要结构
Fig. 2. (a) Difference between the interfacial electrolyte and the bulk electrolyte; (b) main structure of the interfacial electrolyte.

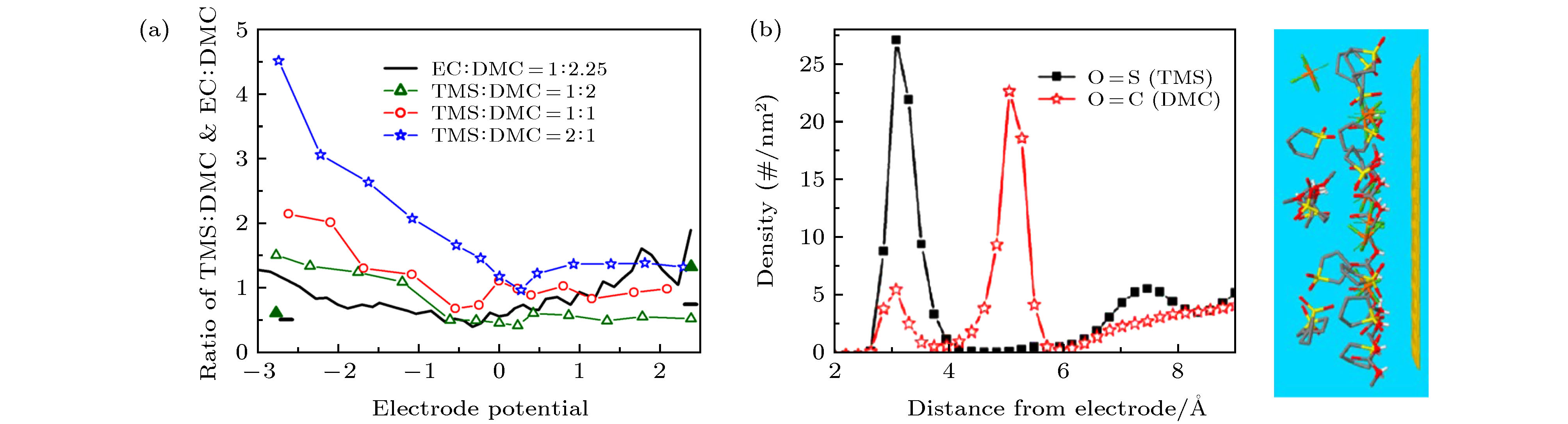
图 4 (a) 界面电解液中TMC:DMC比例与电极电势的关系; (b) 带正电电极界面电解液中TMS和DMC溶剂的微观结构示意图[16]
Fig. 4. (a) Ratio of cumulative densities TMS:DMC in the interfacial electrolyte as a function of electrode potential; (b) schematic diagram of the microstructure of TMS and DMC solvents in the electrolyte at the interface of positively charged electrodes[16]


图 6 (a)
${\rm{PF}}_6^- $ 和共溶剂分子对SN和碳酸酯计算氧化电位的影响; (b) 不同的实验设计验证SN添加剂的作用机理; (c) SN添加剂提高高电压电极/电解液界面稳定性的机理图[22]Fig. 6. (a) Influence of
${\rm{PF}}_6^- $ and co-solvent molecules on the calculated oxidation potential of SN and carbonate; (b) different experimental designs verified the mechanism of SN additive: (c) mechanism of SN additive on improving the stability of high voltage electrode/electrolyte interface[22].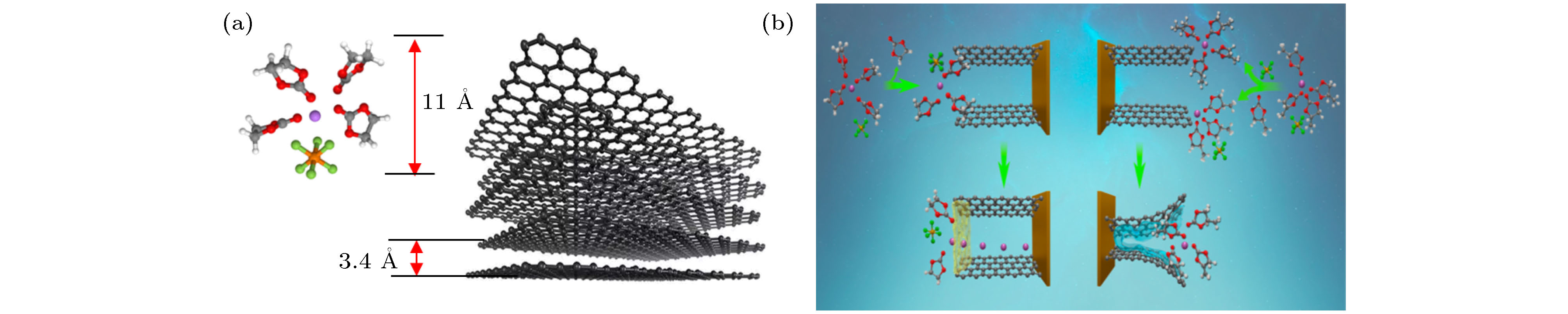
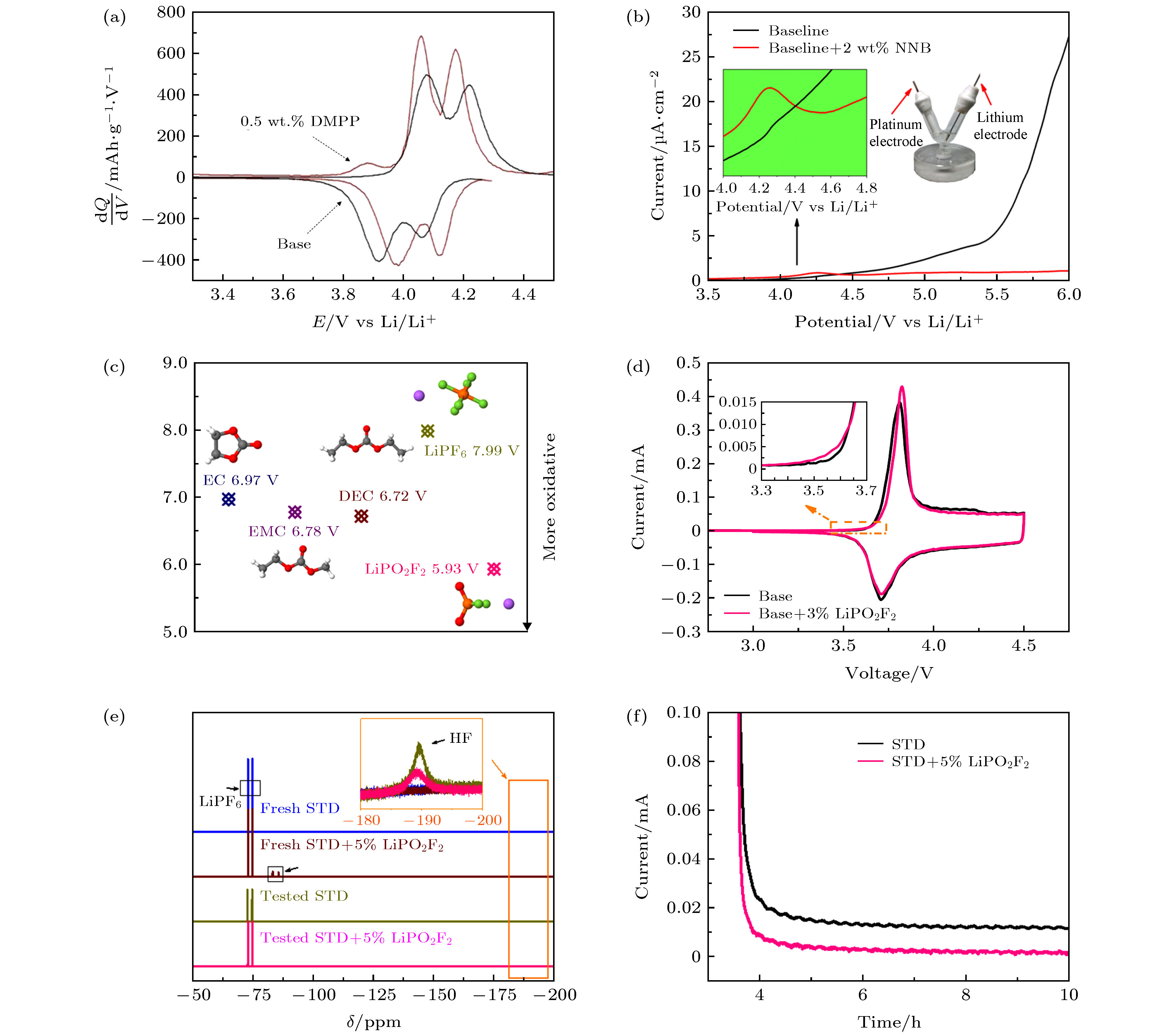
图 8 (a) DMPP添加剂在锰酸锂电极上的C-V曲线; (b) NNB添加剂在Pt电极上的LSV曲线; (c) LiPO2F2与电解液其他组分的计算氧化电位; (d) LiPO2F2添加剂在三元正极上的CV曲线; (e) 电解液在经过电化学测试前后的19F核磁共振谱; (f) 三元正极在不同电解液中的计时电流曲线[31—33]
Fig. 8. (a) C-V curves of DMPP additive on LMO electrode; (b) LSV curves of NNB additive on Pt electrode; (c) calculated oxidation potential (V vs. Li/Li+) of EC, EMC, DEC, LiPF6 and LiPO2F2; (d) C-V curves of LNCM/Li cells with and without LiPO2F2; (e) 19 F NMR spectra of electrolytes before and after electrochemical test; (f) chronoamperometric responses of LNCM/Li cells with and without LiPO2F2[31—33].
-
[1] Nitta N, Wu F, Lee J T, Yushin G 2015 Mater. Today 18 252
Google Scholar
[2] Xu K 2004 Chem. Rev. 104 4303
Google Scholar
[3] Xu K 2014 Chem. Rev. 114 11503
Google Scholar
[4] Winter M, Barnett B, Xu K 2018 Chem. Rev. 118 11433
Google Scholar
[5] Zhang X Q, Chen X, Hou L P, Li B Q, Cheng X B, Huang J Q, Zhang Q 2019 ACS Energy Lett. 4 411
Google Scholar
[6] Chen X, Zhang Q 2020 Acc. Chem. Res. 53 1992
Google Scholar
[7] Winter M 2009 Z. Phys. Chem. 223 1395
Google Scholar
[8] Borodin O, Smith G D 2009 J. Phys. Chem. B 113 1763
Google Scholar
[9] Xing L D, Li W S, Wang C Y, Xu M Q, Tan C L, Yi J 2009 J. Phys. Chem. B 113 16596
[10] Xing L D, Wang C Y, Xu M Q, Li W S, Cai Z P 2009 J. Power Sources 189 689
[11] Xing L D, Wang C Y, Li W S, Xu M Q, Meng X L, Zhao S F 2009 J. Phys. Chem. B 113 5181
[12] Wang Q, Evans N, Zakeeruddin S M, Exnar I, Gra¨tzel M 2007 J. Am. Chem. Soc. 129 3163
[13] Xing L D, Borodin O, Smith G, Li W S 2011 J. Phys. Chem. A 115 13896
Google Scholar
[14] Borodin O, Jow T R 2011 ECS Trans. 33 77
[15] Vatamanu J, Borodin O, Smith G D 2012 J. Phys. Chem. C 116 1114
Google Scholar
[16] Xing L D, Vatamanu J, Bedrov D, Borodin O, Smith G D 2012 J. Phys. Chem. C 116 23871
Google Scholar
[17] Watanabe Y, Kinoshita S I, Wada S, Hoshino K, Morimoto H, Tobishima S I 2008 J. Power Sources 179 770
Google Scholar
[18] Fan X, Ji X, Chen L, Chen J, Deng T, Han F, Yue J, Piao N, Wang R, Zhou X, Xiao X, Chen L, Wang C 2019 Nat. Energy 4 882
Google Scholar
[19] Xing L D, Borodin O 2012 Phys. Chem. Chem. Phys. 14 12838
Google Scholar
[20] Abu-Lebdeh Y, Davidson I 2009 J. Electrochem. Soc. 156 A60
Google Scholar
[21] Kim Y S, Kim T H, Lee H, Song H K 2011 Energy Environ. Sci. 4 4038
Google Scholar
[22] Zhi H Z, Xing L D, Zheng X W, Xu K, Li W S 2017 J. Phys. Chem. Lett. 8 6048
Google Scholar
[23] Wang Y T, Xing L D, Li W S, Bedrov D 2013 J. Phys. Chem. Lett. 4 3992
Google Scholar
[24] Zhuang G V, Yang H, Blizanac B, Ross P N 2015 Electrochem. Solid-State Lett. 8 A441
[25] Tasaki K, Goldberg A, Winter M 2011 Electrochim. Acta 56 10424
Google Scholar
[26] Xing L D, Zheng X W, Schroeder M, Alvarado J, Cresce A W, Xu K 2018 Acc. Chem. Res. 51 282
Google Scholar
[27] Wang C, Xing L D, Vatamanu J, Chen Z, Lan G Y, Li W S, Xu K 2019 Nat. Commun. 10 3423
Google Scholar
[28] Jarry A, Gottis S, Yu Y S, Roque R J, Kim C, Cabana J, Kerr J, Kostecki R 2015 J. Am. Chem. Soc. 137 3533
Google Scholar
[29] Zhan C, Lu J, Kropf A J, Wu T P, Jansen A N, Sun Y K, Qiu X P, Amine K 2013 Nat. Commun. 4 2437
Google Scholar
[30] Wang K, Xing L D, Xu K, Zhou H B, Li W S 2019 ACS Appl. Mater. Interfaces 11 31490
Google Scholar
[31] Zhu Y M, Rong H B, Mai S W, Luo X Y, Li X P, Li W S 2015 J. Power Sources 299 485
Google Scholar
[32] Zheng Q F, Xing L D, Yang X R, Li X F, Ye C C, Wang K, Huang Q M, Li W S 2018 ACS Appl. Mater. Interfaces 10 16843
Google Scholar
[33] Chen J W, Xing L D, Yang X R, Liu X, Li T J, Li W S 2018 Electrochim. Acta 290 568
Google Scholar

 下载:
下载:


计量
- 文章访问数: 29684
- PDF下载量: 1262
- 被引次数: 0
