










-
电离能是原子和分子的重要的特性参数, 在光物理和光化学过程中起着重要作用, 精确电离能对相关研究具有重要意义. 电离能是调试零动能光谱信号的重要参考数据, 在判断异构物数量和分子构型方面也起着关键作用. 1,3-二乙氧基苯是一种重要的苯的衍生物, 实验证实在超声分子束中包含两种旋转异构物I(down-up)和III(down-down). 它们的精确电离能还未见文献报道. 本文采用直线式飞行时间质谱仪测量了静电场中1,3-二乙氧基苯光电离效率曲线, 通过不同电场强度下测量的电离能(Stark效应)对场强的平方根线性拟合给出了两种异构物I和III精确的电离能分别为(62419 ± 2) cm–1和(63378 ± 2) cm–1. 相对于通常的脉冲电场加速机制和零动能光谱测量的电离能, 精确度大约分别由(± 10) cm–1和(± 5) cm–1提高到(± 2) cm–1. 分析和讨论了不同方法测量的物理机制和优缺点.
-
关键词:
- 电离能 /
- 光电离效率 /
- Stark效应 /
- 1, 3-二乙氧基苯
Ionization energy (IE) is an important characteristic parameter of atoms or molecules. It plays an important role in the process of photophysics and photochemistry. The precise ionization energy is very important for relevant research. Especially, it is very useful for adjusting the signal of the zero-kinetic energy (ZEKE) spectrum, and it also plays a key role in judging the number of rotamers and molecular configuration. In linear time-of-flight mass spectrometers, pulsed electric fields are usually used to drive photo-ionized ions to the detector to produce the photoionization efficiency (PIE) spectrum. The ionization energy is directly obtained from the PIE curve. The uncertainty of the measured IE is usually greater than or equal to ± 10 cm–1. The ZEKE spectroscopy is based on the long-lived Rydberg state field ionization technology. In the ZEKE experiments, the laser excites molecules to the Rydberg state and then a pulsed field ionization (PFI) is used for measurement. A peak with high signal-to-noise ratio and narrow linewidth signal appears near the ionization threshold. Therefore, the more accurate ionization energy can be obtained, and the uncertainty of the measured value is about ± 5 cm–1. The 1,3-diethoxybenzene is an important benzene derivative, and experiments have confirmed that there are two rotamers, i.e. I (down-up) and III (down-down) in the supersonic molecular beam. In this paper, a linear time-of-flight mass spectrometer is used to measure the photoionization efficiency curves of 1,3-diethoxybenzene in electrostatic fields. From the linear fitting of the ionization energy values measured under different electric fields (Stark effect) to the square root of the field strengths, the precise ionization energy values of rotamer I and rotamer III are determined to be (62419 ± 2) cm–1 and (63378 ± 2) cm–1, respectively. Compared with the accuracies of the values measured by the usual pulsed electric field acceleration mechanism and the ZEKE spectroscopy, the accuracy is improved from about ± 10 and ± 5 to ± 2 cm–1, respectively. The physical mechanism, advantages and disadvantages of different methods are analyzed and discussed. The present research results show that the ionization energy measured in the electrostatic field is more accurate, the physical meaning of the measurement process is clear, and the threshold data are easy to collect. This is the first report on the precise ionization energy of 1,3-diethoxybenzene rotamers.-
Keywords:
- ionization energy /
- photoionization efficiency /
- Stark effect /
- 1,3-diethoxybenzene
[1] Zhang L J, Yu D, Dong C W, Cheng M, Hu L L, Zhou Z M, Du Y K, Zhu Q H, Zhang C H 2013 Spectrochim. Acta, Part A 104 235
 Google Scholar
Google Scholar
[2] Yang S C, Huang H W, Tzeng W B 2010 J. Phys. Chem. A 114 11144
Google Scholar
[3] Qin C, Tzeng S Y, Zhang B, Tzeng W B 2019 J. Mol. Spectrosc. 355 26
Google Scholar
[4] Xu Y Q, Tzeng S Y, Zhang B, Tzeng W B 2013 Spectrochim. Acta, Part A 102 365
Google Scholar
[5] Lee Y R, Kim M H, Kim H L, Kwon C H 2018 J. Chem. Phys. 149 174302
Google Scholar
[6] Wu P Y, Tzeng W B 2015 J. Mol. Spectrosc. 316 72
Google Scholar
[7] Tsai C Y, Tzeng W B 2013 J. Photoch. Photobio. A 270 53
Google Scholar
[8] Dai W S, Zhang Z, Du Y K 2020 Spectrochim. Acta, Part A 224 117398
Google Scholar
[9] Xiao D Q, Yu D, Xu X L, Yu Z J, Cheng M, Du Y K, Zheng W J, Zhu Q H 2009 Phys. Chem. Chem. Phys. 11 3532
Google Scholar
[10] Xu Y Q, Tzeng S Y, Shivatare V, Takahashi K, Zhang B, Tzeng W B 2015 J. Chem. Phys. 142 124314
Google Scholar
[11] Huang J H, Huang K L, Liu S Q, Luo Q, Tzeng W B 2007 J. Photoch. Photobio. A 188 252
Google Scholar
[12] Li C Y, Lin J L, Tzeng W B 2005 J. Chem. Phys. 122 044311
Google Scholar
[13] Zhang L J, Dong C W, Cheng M, Hu L L, Du Y K, Zhu Q H, Zhang C H 2012 Spectrochim. Acta, Part A 96 578
Google Scholar
[14] Li C Y, Pradhan M, Tzeng W B 2005 Chem. Phys. Lett. 411 506
Google Scholar
[15] Lin J L, Li C Y, Tzeng W B 2004 J. Chem. Phys. 120 10513
Google Scholar
[16] Qin C, Tzeng S Y, Zang B, Tzeng W B 2014 Acta Phys. Chim. Sin. 30 1416
Google Scholar
[17] Wu P Y, Tzeng S Y, Hsu Y C, Tzeng W B 2017 J. Mol. Spectrosc. 332 3
Google Scholar
[18] Lin J L, Tzeng W B 2000 Phys. Chem. Chem. Phys 2 3759
Google Scholar
[19] Shivatare V, Tzeng W B 2014 Bull. Korean Chem. Soc. 35 815
Google Scholar
[20] Lin J L, Li Y C, Tzeng W B 2007 Chem. Phys. 334 189
Google Scholar
[21] Ketkov S Y, Tzeng S Y, Wu P Y, Markin G V, Tzeng W B 2017 Chem. Eur. J. 23 1
Google Scholar
[22] Zhang L J, Li D Z, Cheng M, Du Y K, Zhu Q H 2017 Spectrochim. Acta, Part A 183 177
Google Scholar
[23] Hao J Y, Duan C Y, Yang Y G, Li C Y, Jia S T 2020 J. Mol. Spectrosc. 369 111258
Google Scholar
[24] Jin Y H, Zhao Y, Yang Y G, Wang L R, Li C Y, Jia S T 2018 Chem. Phys. Lett. 692 395
Google Scholar
[25] 李鑫, 赵岩, 靳颖辉, 王晓锐, 余谢秋, 武媚, 韩昱行, 杨勇刚, 李昌勇, 贾锁堂 2017 物理学报 66 093301
Google Scholar
Li X, Zhao Y, Jin Y H, Wang X R, Yu X Q, Wu M, Han Y Y, Yang Y G, Li C Y, Jia S T 2017 Acta Phys. Sin. 66 093301
Google Scholar
[26] Zhao Y, Jin Y H, Hao J Y, Yang Y G, Wang L R, Li C Y, Jia S T 2019 Spectrochim. Acta, Part A 207 328
Google Scholar
[27] Ullrich S, Geppert W D, Dessent C E H, Mu1ler-Dethlefs K 2000 J. Phys. Chem. A 104 11864
[28] Wilke M, Schneider M, Wilke J, Ruiz-Santoyo J A, Campos-Amador J J, Gonzalez-Medina M. E, Alvarez-Valtierra L, Schmitt M 2017 J. Mol. Struct. 1140 59
Google Scholar
[29] 李昌勇, 张临杰, 赵建明, 贾锁堂 2012 物理学报 61 163202
Google Scholar
Li C Y, Zhang L J, Zhao J M, Jia S T 2012 Acta Phys. Sin. 61 163202
Google Scholar
[30] Li C Y, Hao T, Zgang H, Zhu X B, TAO G Q, Zhang L J, Zhao J M, Jia S T 2012 J. Phys. Soc. Jpn. 81 044302
Google Scholar
[31] Dong H, Wang T, Li C Y, Zhao J M, Zhang L J 2013 Chin. Phys. B 22 073201
Google Scholar
[32] Dong H, Hang K S, Li C Y, Zhao J M, Zhang L, Jia S T 2014 Chin. Phys. B 23 093202
Google Scholar
[33] 董慧杰, 王新宇, 李昌勇, 贾锁堂 2015 物理学报 64 093201
Google Scholar
Gong H J, Wang X Y, Li C Y, Jia S T 2015 Acta Phys. Sin. 64 093201
Google Scholar
[34] Wang L M, Li C Y, Zhang H, Zhang L J, Yang Y G, Man Y, Zhao J M, Jia S T 2016 Phys. Rev. A 93 033416
Google Scholar
[35] Chupka W A 1993 J. Chem. Phys. 98 4520
Google Scholar
[36] Zhang B, Li C Y, Su H W, Lin J L, Tzeng W B 2004 Chem. Phys. Lett. 390 65
Google Scholar
[37] Choi K W, Choi S, Baek S J, Kim S K 2007 J. Chem. Phys. 126 034308
Google Scholar
-
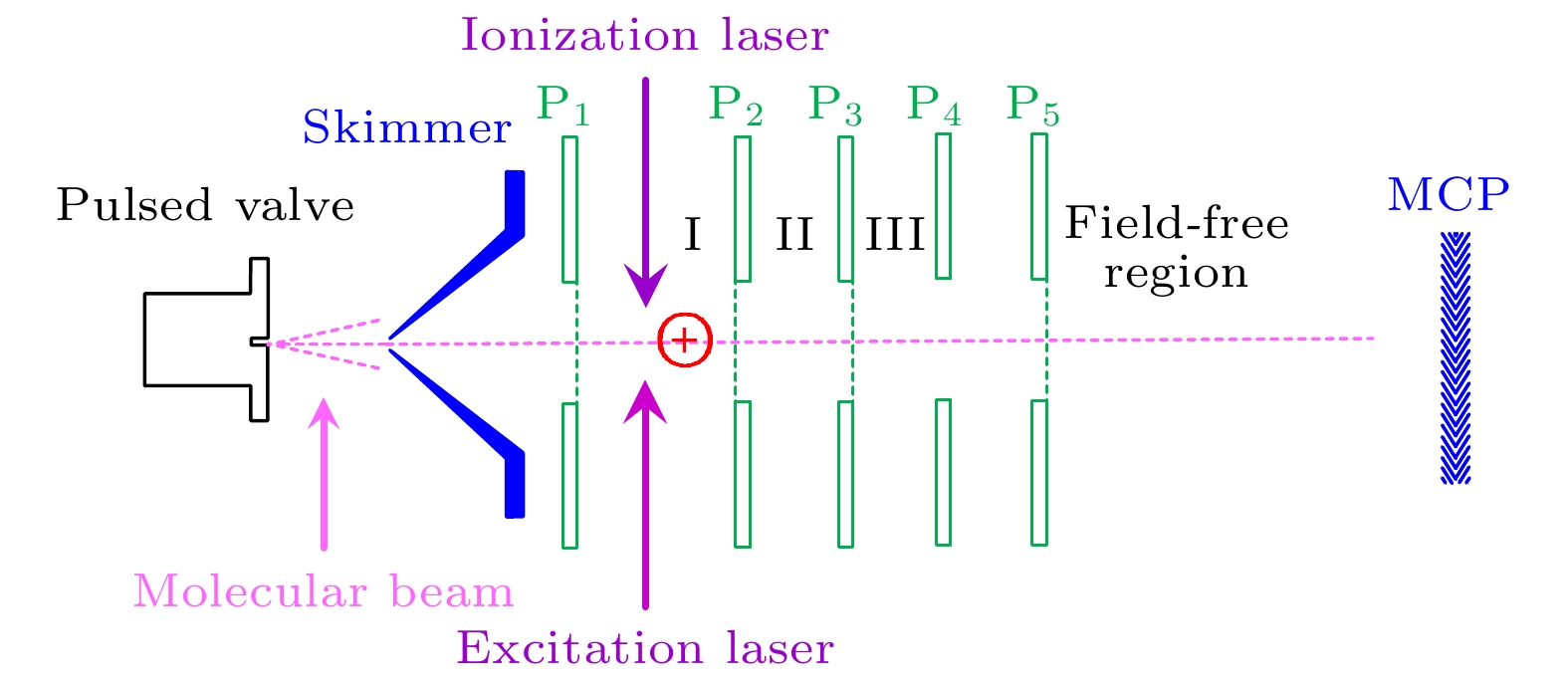
图 1 直线式飞行时间质谱仪原理图. P1, P2, P3, P4, P5为离子透镜的片状电极; P1与P2间区域I为激光和分子束相互作用区. MCP为微通道板探测器
Fig. 1. Schematic diagram of a linear time-of-flight mass spectrometer. P1, P2, P3, P4, P5 are the electrodes of the electrostatic lens; the region I between P1 and P2 is the interaction area between lasers and molecular beam. MCP is a microchannel plate detector.
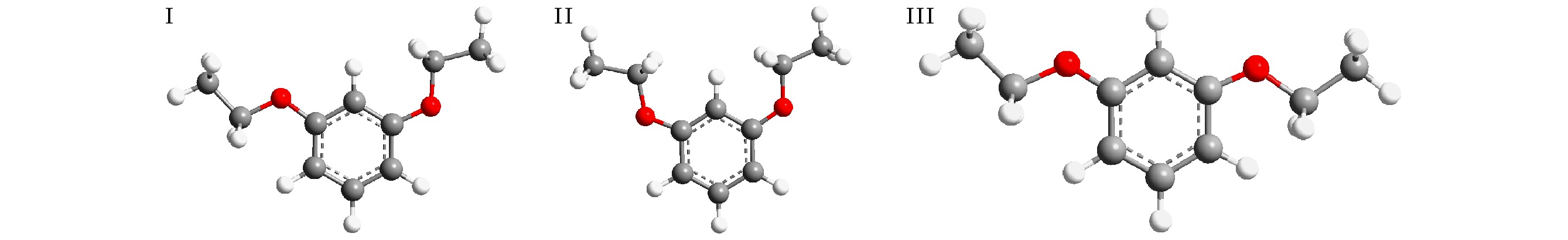
图 2 1,3-二乙氧基苯分子的三种构型, 根据取代基OC2H5的方向命名三种构型分别为I, down-up; II, up-up; III, down-down
Fig. 2. Three configurations of 1,3-diethoxybenzene molecule. The three configurations are named according to the direction of the substituent OC2H5 as I, down-up; II, up-up; III, down-down.
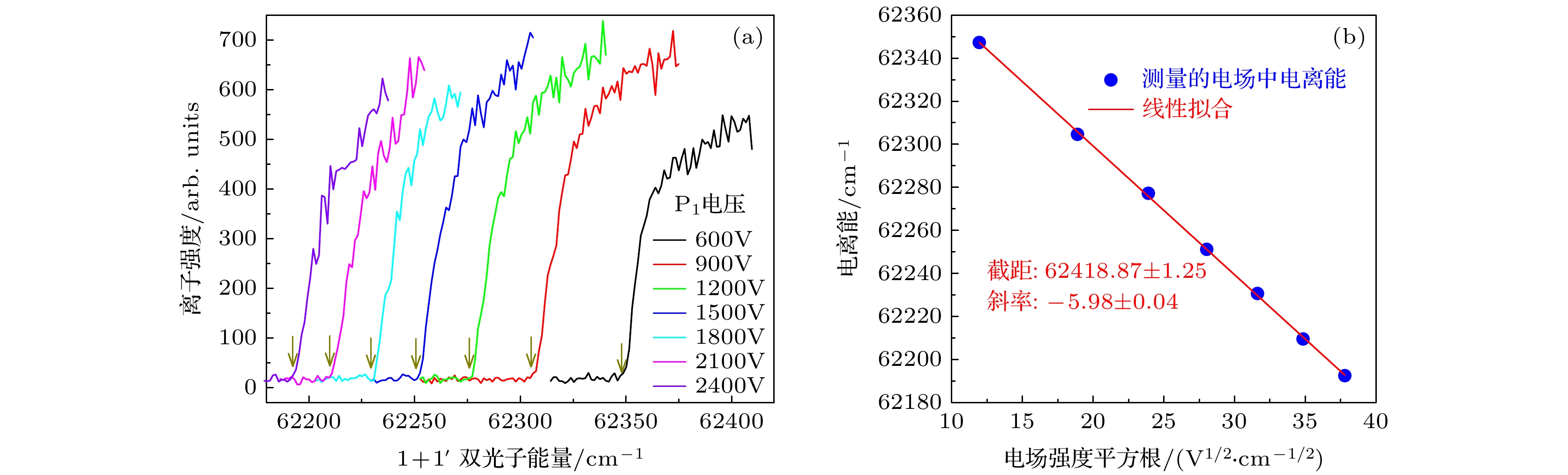
图 3 异构物I在不同电场中的光电离效率曲线(a)及其测量的电离能对电场强度平方根的线性拟合(b). 图(a)中箭头指向了电场中电离阈值的取值点
Fig. 3. The photoionization efficiency curves of isomer I (down-up) in different electric fields (a), and the linear fitting of the measured ionization energy to the square root of the electric field intensity (b). The arrows in figure (a) point to the ionization thresholds in the electric fields.
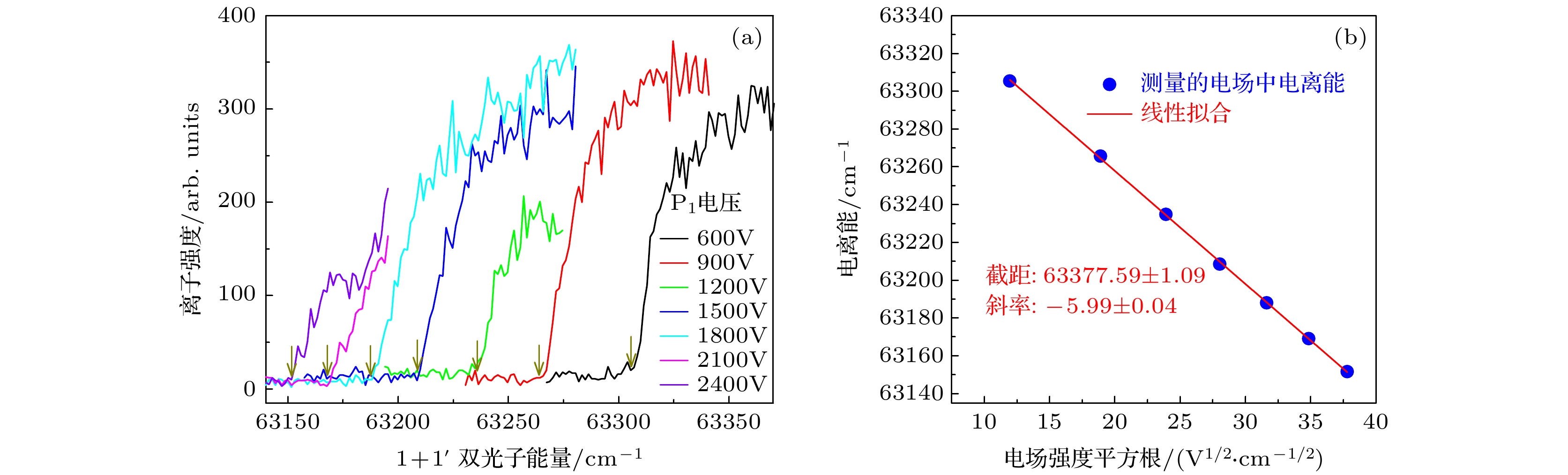
图 4 异构物III在不同电场中的光电离效率曲线(a)及其测量的电离能对电场强度平方根的线性拟合(b). 图(a)中箭头指向了电场中电离阈值的采集点
Fig. 4. The photoionization efficiency curves of isomer III (down-down) in different electric fields (a), and the linear fitting of the measured ionization energy to the square root of the electric field intensity (b). The arrows in figure (a) point to the ionization thresholds in the electric fields.
-
[1] Zhang L J, Yu D, Dong C W, Cheng M, Hu L L, Zhou Z M, Du Y K, Zhu Q H, Zhang C H 2013 Spectrochim. Acta, Part A 104 235
Google Scholar
[2] Yang S C, Huang H W, Tzeng W B 2010 J. Phys. Chem. A 114 11144
Google Scholar
[3] Qin C, Tzeng S Y, Zhang B, Tzeng W B 2019 J. Mol. Spectrosc. 355 26
Google Scholar
[4] Xu Y Q, Tzeng S Y, Zhang B, Tzeng W B 2013 Spectrochim. Acta, Part A 102 365
Google Scholar
[5] Lee Y R, Kim M H, Kim H L, Kwon C H 2018 J. Chem. Phys. 149 174302
Google Scholar
[6] Wu P Y, Tzeng W B 2015 J. Mol. Spectrosc. 316 72
Google Scholar
[7] Tsai C Y, Tzeng W B 2013 J. Photoch. Photobio. A 270 53
Google Scholar
[8] Dai W S, Zhang Z, Du Y K 2020 Spectrochim. Acta, Part A 224 117398
Google Scholar
[9] Xiao D Q, Yu D, Xu X L, Yu Z J, Cheng M, Du Y K, Zheng W J, Zhu Q H 2009 Phys. Chem. Chem. Phys. 11 3532
Google Scholar
[10] Xu Y Q, Tzeng S Y, Shivatare V, Takahashi K, Zhang B, Tzeng W B 2015 J. Chem. Phys. 142 124314
Google Scholar
[11] Huang J H, Huang K L, Liu S Q, Luo Q, Tzeng W B 2007 J. Photoch. Photobio. A 188 252
Google Scholar
[12] Li C Y, Lin J L, Tzeng W B 2005 J. Chem. Phys. 122 044311
Google Scholar
[13] Zhang L J, Dong C W, Cheng M, Hu L L, Du Y K, Zhu Q H, Zhang C H 2012 Spectrochim. Acta, Part A 96 578
Google Scholar
[14] Li C Y, Pradhan M, Tzeng W B 2005 Chem. Phys. Lett. 411 506
Google Scholar
[15] Lin J L, Li C Y, Tzeng W B 2004 J. Chem. Phys. 120 10513
Google Scholar
[16] Qin C, Tzeng S Y, Zang B, Tzeng W B 2014 Acta Phys. Chim. Sin. 30 1416
Google Scholar
[17] Wu P Y, Tzeng S Y, Hsu Y C, Tzeng W B 2017 J. Mol. Spectrosc. 332 3
Google Scholar
[18] Lin J L, Tzeng W B 2000 Phys. Chem. Chem. Phys 2 3759
Google Scholar
[19] Shivatare V, Tzeng W B 2014 Bull. Korean Chem. Soc. 35 815
Google Scholar
[20] Lin J L, Li Y C, Tzeng W B 2007 Chem. Phys. 334 189
Google Scholar
[21] Ketkov S Y, Tzeng S Y, Wu P Y, Markin G V, Tzeng W B 2017 Chem. Eur. J. 23 1
Google Scholar
[22] Zhang L J, Li D Z, Cheng M, Du Y K, Zhu Q H 2017 Spectrochim. Acta, Part A 183 177
Google Scholar
[23] Hao J Y, Duan C Y, Yang Y G, Li C Y, Jia S T 2020 J. Mol. Spectrosc. 369 111258
Google Scholar
[24] Jin Y H, Zhao Y, Yang Y G, Wang L R, Li C Y, Jia S T 2018 Chem. Phys. Lett. 692 395
Google Scholar
[25] 李鑫, 赵岩, 靳颖辉, 王晓锐, 余谢秋, 武媚, 韩昱行, 杨勇刚, 李昌勇, 贾锁堂 2017 物理学报 66 093301
Google Scholar
Li X, Zhao Y, Jin Y H, Wang X R, Yu X Q, Wu M, Han Y Y, Yang Y G, Li C Y, Jia S T 2017 Acta Phys. Sin. 66 093301
Google Scholar
[26] Zhao Y, Jin Y H, Hao J Y, Yang Y G, Wang L R, Li C Y, Jia S T 2019 Spectrochim. Acta, Part A 207 328
Google Scholar
[27] Ullrich S, Geppert W D, Dessent C E H, Mu1ler-Dethlefs K 2000 J. Phys. Chem. A 104 11864
[28] Wilke M, Schneider M, Wilke J, Ruiz-Santoyo J A, Campos-Amador J J, Gonzalez-Medina M. E, Alvarez-Valtierra L, Schmitt M 2017 J. Mol. Struct. 1140 59
Google Scholar
[29] 李昌勇, 张临杰, 赵建明, 贾锁堂 2012 物理学报 61 163202
Google Scholar
Li C Y, Zhang L J, Zhao J M, Jia S T 2012 Acta Phys. Sin. 61 163202
Google Scholar
[30] Li C Y, Hao T, Zgang H, Zhu X B, TAO G Q, Zhang L J, Zhao J M, Jia S T 2012 J. Phys. Soc. Jpn. 81 044302
Google Scholar
[31] Dong H, Wang T, Li C Y, Zhao J M, Zhang L J 2013 Chin. Phys. B 22 073201
Google Scholar
[32] Dong H, Hang K S, Li C Y, Zhao J M, Zhang L, Jia S T 2014 Chin. Phys. B 23 093202
Google Scholar
[33] 董慧杰, 王新宇, 李昌勇, 贾锁堂 2015 物理学报 64 093201
Google Scholar
Gong H J, Wang X Y, Li C Y, Jia S T 2015 Acta Phys. Sin. 64 093201
Google Scholar
[34] Wang L M, Li C Y, Zhang H, Zhang L J, Yang Y G, Man Y, Zhao J M, Jia S T 2016 Phys. Rev. A 93 033416
Google Scholar
[35] Chupka W A 1993 J. Chem. Phys. 98 4520
Google Scholar
[36] Zhang B, Li C Y, Su H W, Lin J L, Tzeng W B 2004 Chem. Phys. Lett. 390 65
Google Scholar
[37] Choi K W, Choi S, Baek S J, Kim S K 2007 J. Chem. Phys. 126 034308
Google Scholar

 下载:
下载:
计量
- 文章访问数: 8207
- PDF下载量: 89
- 被引次数: 0
