










-
Lithium-oxygen battery possesses an extremely high theoretical energy density (
$ \approx$ 3500 W·h·kg–1), and is an ideal next-generation energy storage system. The ideal operation of lithium-oxygen batteries is based on the electrochemical formation (discharge) and decomposition (charge) of lithium peroxide (Li2O2). At the beginning of the discharge, oxygen is reduced on the electrode, forming an oxygen radical (${\rm O}^{-}_{2} $ ). The$ {\rm O}^{-}_{2}$ successively combines with an Li ion, forming the metastable LiO2. The LiO2 may subsequently undergo two different reaction pathways: a chemical disproportionation and a continuous electrochemical reduction, thereby resulting in the formation of Li2O2. Therefore, the oxygen reduction reaction (ORR) is an important step in the discharge process. Studies have shown that graphene is considered as the most promising cathode material for non-aqueous lithium-oxygen batteries. Moreover, it is found that nitrogen-doped graphene has higher electrocatalytic activity than intrinsic graphene for the ORR. However, up to now, the mechanism of improving the ORR for nitrogen-doped graphene is still unclear, and the effects of different N-doping concentrations on the ORR have not been reported. In this work, on the basis of the first-principles calculations, the reduction mechanism of O2 molecule by nitrogen-doped graphene with different N concentrations is studied. Results show that after doping N atoms, the adsorption energy of O2 molecules increases, the O—O bond length is elongated, and the transferred charge increases, which indicates that nitrogen-doped graphene enhances the reduction ability of O2 molecule. Bader charge analysis shows that both N atom and O2 molecule obtain charges from C atom, and N atom also provides charges for O2 molecule, which is consistent with the electronegativity of carbon, nitrogen and oxygen. This charge transfer results in the stronger interaction between the O2 molecule and the substrate, and can reveal the reason why nitrogen-doped graphene can improve the ORR. In addition, it is found that the reduction ability of O2 molecule is best when the N-doping ratio is 3.13 at%. It is hoped that this work will play a guiding role in the synthesizing the nitrogen-doped graphene materials, and will be helpful in optimizing the cathode materials of lithium-oxygen batteries.-
Keywords:
- lithium air batteries /
- N-doped graphene /
- oxygen molecule adsorption /
- oxygen reduction mechanism /
- first-principles
 128801-Suppl.pdf
128801补充材料图S1—图S7
128801-Suppl.pdf
128801补充材料图S1—图S7
[1] Yang Z, Zhang J, Kintner-Meyer M C W, Lu X, Choi D, Lemmon J P, Liu J 2011 Chem. Rev. 111 3577
 Google Scholar
Google Scholar
[2] Kwak W J, Kim H, Jung H G, Aurbach D, Sun Y K 2018 J. Electrochem. Soc. 165 A2274
Google Scholar
[3] Zhao N, Li C, Guo X 2014 Energy Technol. 2 317
Google Scholar
[4] Wen Z, Shen C, Lu Y 2015 ChemPlusChem 80 270
Google Scholar
[5] Lim H D, Lee B, Bae Y, Park H, Ko Y, Kim H, Kim J, Kang K 2017 Chem. Soc. Rev. 46 2873
Google Scholar
[6] Dai W, Cui X, Zhou Y, Zhao Y, Wang L, Peng L, Chen W 2019 Small Methods 3 1800358
Google Scholar
[7] Lu J, Li L, Park J B, Sun Y K, Wu F, Amine K 2014 Chem. Rev. 114 5611
Google Scholar
[8] Aurbach D, McCloskey B D, Nazar L F, Bruce P G 2016 Nat. Energy 1 1
[9] Abraham K M, Jiang Z 1996 Electrochem. Sci. Technol. 143 1
Google Scholar
[10] Ogasawara T, Débart A l, Holzapfel M, Novák P, Bruce P G 2006 J. Am. Chem. Soc. 128 1390
Google Scholar
[11] Wang Y, Lai N C, Lu Y R, Zhou Y, Dong C L, Lu Y C 2018 Joule 2 2364
Google Scholar
[12] Lu Y, Tong S, Qiu F, Jiang J, Feng N, Zhang X, He P, Zhou H 2016 J. Power Sources 329 525
Google Scholar
[13] Liu C J, Brant W R, Younesi R, Dong Y Y, Edstrom K, Gustafsson T, Zhu J F 2017 ChemSusChem 10 1592
Google Scholar
[14] Cui Y M, Wen Z Y, Liang X, Lu Y, Jin J, Wu M F, Wu X W 2012 Energy Environ. Sci. 5 7893
Google Scholar
[15] Wang J J, Li Y L, Sun X L 2013 Nano Energy 2 443
Google Scholar
[16] Ma Z, Yuan X, Li L, Ma Z F, Wilkinson D P, Zhang L, Zhang J 2015 Energy Environ. Sci. 8 2144
Google Scholar
[17] Tang Y, Qiao H, Wang H, Tao P 2013 J. Mater. Chem. A 1 12512
Google Scholar
[18] Xiao J, Mei D, Li X, Xu W, Wang D, Graff G L, Bennett W D, Nie Z, Saraf L V, Aksay I A, Liu J, Zhang J G 2011 Nano Lett. 11 5071
Google Scholar
[19] Jin L, Xu L, Morein C, Chen C, Lai M, Dharmarathna S, Dobley A, Suib S L 2010 Adv. Funct. Mater. 20 3373
Google Scholar
[20] Gao R, Shang Z, Zheng L, Wang J, Sun L, Hu Z, Liu X 2019 Inorg. Chem 58 4989
Google Scholar
[21] Park J B, Lee S H, Jung H G, Aurbach D, Sun Y K 2018 Adv. Mater. 30 1704162
Google Scholar
[22] Liu S, Wang J, Zeng J, Ou J, Li Z, Liu X, Yang S 2010 J. Power Sources 195 4628
Google Scholar
[23] Soin N, Roy S S, Lim T H, McLaughlin J A D 2011 Mater. Chem. Phys. 129 1051
Google Scholar
[24] Li Y, Wang J, Li X, Geng D, Li R, Sun X 2011 Chem. Commun. 47 9438
Google Scholar
[25] Sun B, Wang B, Su D, Xiao L, Ahn H, Wang G 2012 Carbon 50 727
Google Scholar
[26] Yang Y, Shi M, Zhou Q F, Li Y S, Fu Z W 2012 Electrochem. Commun. 20 11
Google Scholar
[27] Kim H, Lim H D, Kim J, Kang K 2014 J. Mater. Chem. A 2 33
Google Scholar
[28] Jung H G, Jeong Y S, Park J B, Sun Y K, Scrosati B, Lee Y J 2013 ACS Nano 7 3532
Google Scholar
[29] Shao Y, Zhang S, Engelhard M H, Li G, Shao G, Wang Y, Liu J, Aksay I A, Lin Y 2010 J. Mater. Chem. 20 7491
Google Scholar
[30] Li Y, Wang J, Li X, Liu J, Geng D, Yang J, Li R, Sun X 2011 Electrochem. Commun. 13 668
Google Scholar
[31] Li Y, Wang J, Li X, Geng D, Banis M N, Li R, Sun X 2012 Electrochem. Commun. 18 12
Google Scholar
[32] Higgins D, Chen Z, Lee D U, Chen Z 2013 J. Mater. Chem. A 1 2639
Google Scholar
[33] Zhao C, Yu C, Liu S, Yang J, Fan X, Huang H, Qiu J 2015 Adv. Funct. Mater. 25 6913
Google Scholar
[34] Yan H J, Xu B, Shi S Q, Ouyang C Y 2012 J. Appl. Phys. 112 104316
Google Scholar
[35] Kresse G, Hafner J 1993 Phys. Rev. B 47 558
Google Scholar
[36] Kresse G, Furthmüller J 1996 Phys. Rev. B 54 11169
Google Scholar
[37] Kresse G, Joubert D 1999 Phys. Rev. B 59 1758
[38] Blöchl P E 1994 Phys. Rev. B 50 17953
Google Scholar
[39] Perdew J P, Burke K, Ernzerhof M 1996 Phys. Rev. Lett. 77 3865
Google Scholar
[40] Perdew J P, Ernzerhof M, Burke K 1996 J. Chem. Phys. 105 9982
Google Scholar
[41] Grimme S 2006 J. Comput. Chem. 27 1787
Google Scholar
[42] Monkhorst H J, Pack J D 1976 Phys. Rev. B 13 5188
Google Scholar
[43] Pramanik A, Kang H S 2011 J. Phys. Chem. C 115 10971
Google Scholar
[44] Novoselov K S, Geim A K, Morozov S V, Jiang D, Zhang Y, Dubonos S V, Grigorieva I V, Firsov A A 2004 Science 306 666
Google Scholar
[45] Avouris P, Chen Z, Perebeinos V 2007 Nature Nanotech. 2 605
Google Scholar
[46] Tang W, Sanville E, Henkelman G 2009 J. Phys.: Condens. Matter 21 084204
Google Scholar
-

图 1 本征石墨烯的几何结构与电子性质 (a)结构图; (b)能带和态密度图; (c)布里渊区高对称点的示意图
Fig. 1. Optimized structure and electronic properties of graphene: (a) Optimized structure; (b) the energy band and density of states (DOS); (c) the irreducible Brillouin zone. The irreducible k-point path ΓMKΓ corresponds to the graphene.
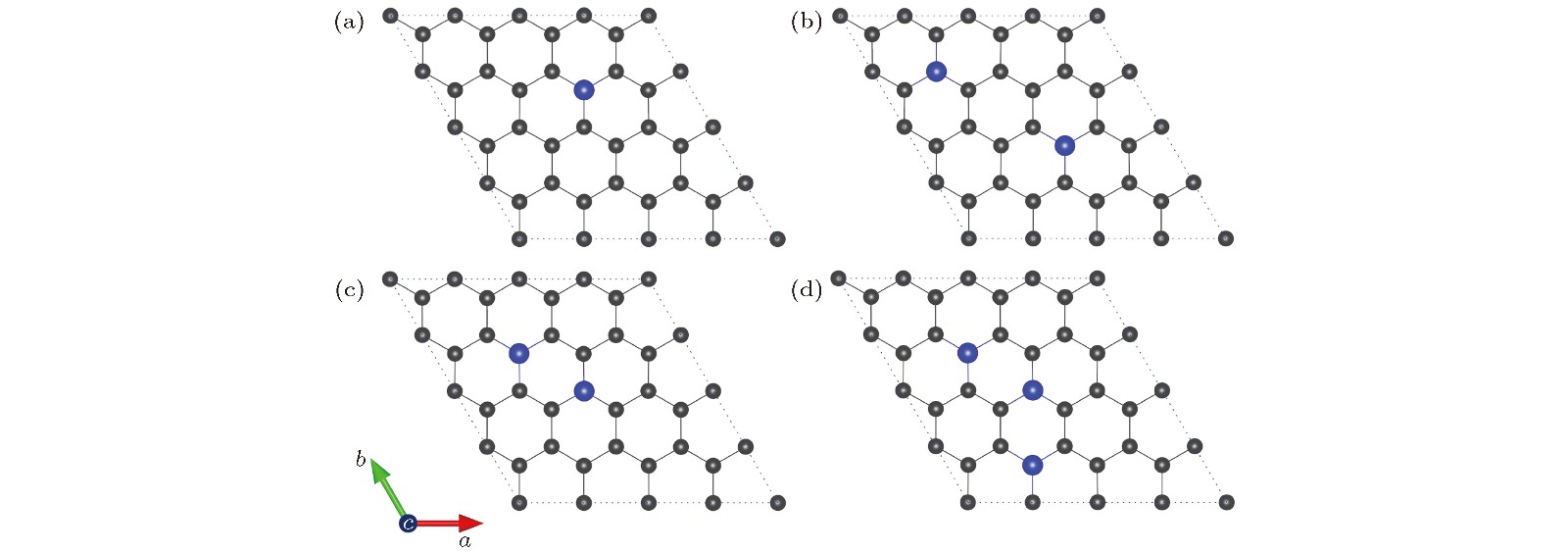
图 2 NxG (x = 1, 2, 3)的最稳定构型(碳原子和氮原子分别用灰色和蓝色小球表示) (a) N1G (x = 1); (b) N2G-1 (x = 2); (c) N2G-2 (x = 2, 亚稳态构型); (d) N3G (x = 3)
Fig. 2. Optimal structures of NxG (x = 1, 2, 3): (a) N1G (x = 1); (b) N2G-1 (x = 2); (c) N2G-2 (x = 2, metastable state); (d) N3G (x = 3). The C atom and N atom are represented by grey and blue sphere, respectively.
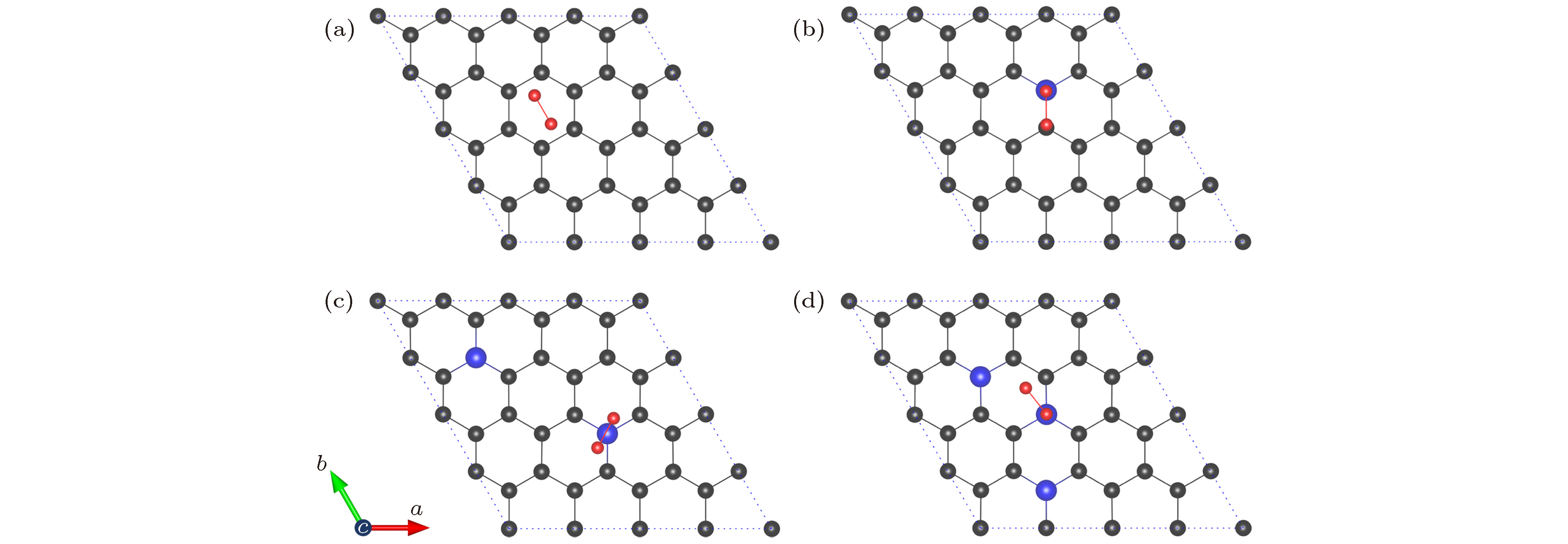
图 3 NxG (x = 0, 1, 2, 3) 吸附氧分子的最稳定构型(碳原子、氮原子和氧原子分别用灰色、蓝色和红色小球表示) (a) G-O2; (b) N1G-O2; (c) N2G-O2; (d) N3G-O2
Fig. 3. Optimal structures of NxG (x = 0, 1, 2, 3): (a) G-O2; (b) N1G-O2; (c) N2G-O2; (d) N3G-O2. The C atom, N atom, and O atom are represented by grey, blue, and red spheres, respectively.
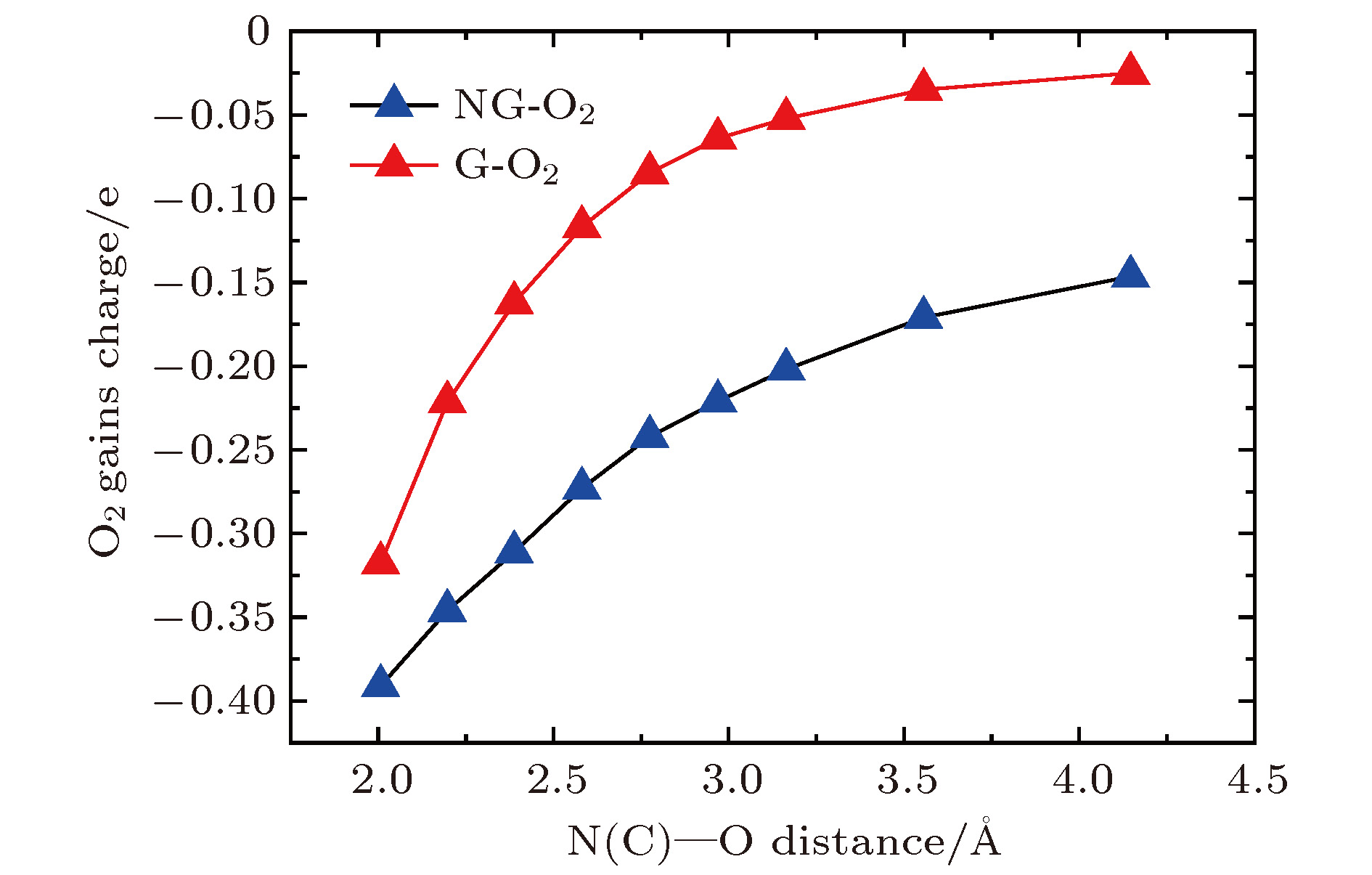
图 4 G-O2和N1G-O2中氧分子获得的电荷与C/N-O距离之间的关系
Fig. 4. Charges of O2 molecule vs. the C-O distance in the G-O2 system (red line) and the N-O distance in the N1G-O2 system (blue line).
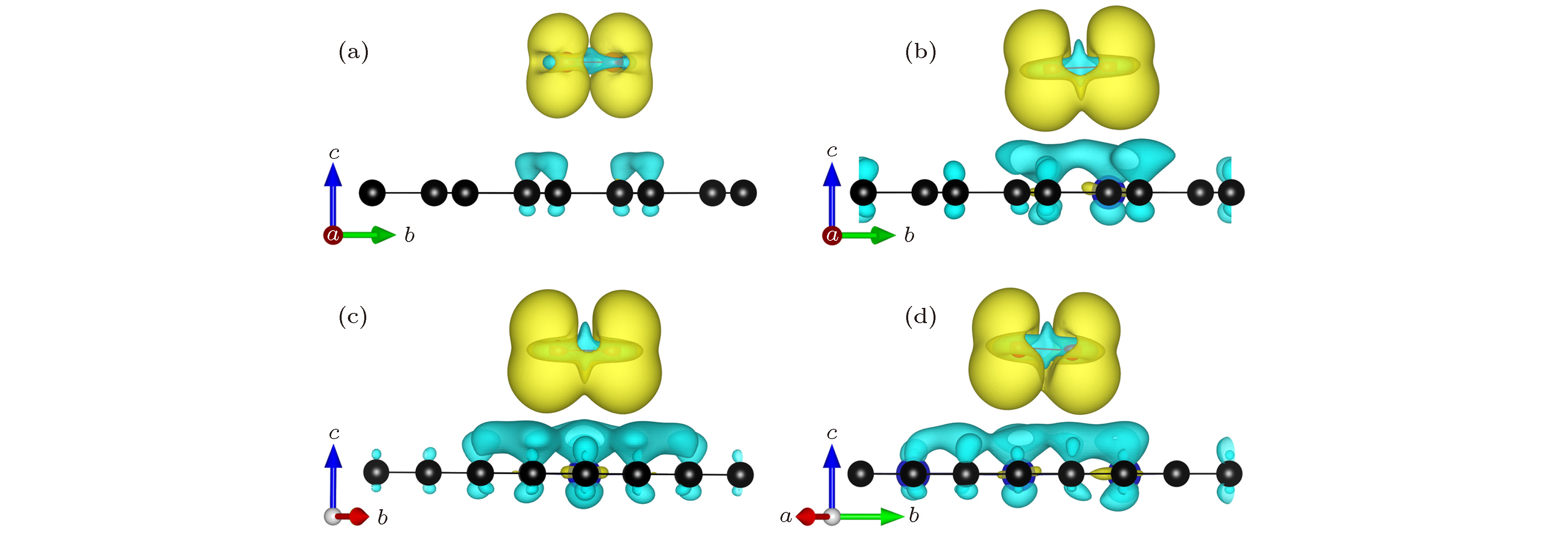
图 5 NxG-O2 (x = 0, 1, 2, 3) 差分电荷密度图(黄色代表电荷聚集, 蓝色代表电荷失去; 电荷等势面是6 × 10–4 e/Å3) (a) G-O2; (b) N1G-O2; (c) N2G-O2; (d) N3G-O2
Fig. 5. Charge density difference of NxG-O2 (x = 0, 1, 2, 3): (a) G-O2; (b) N1G-O2; (c) N2G-O2; (d) N3G-O2. The gain of electrons is indicated in yellow and the loss of electrons is indicated in blue. The isosurface value is 6 × 10–4 e/Å3.
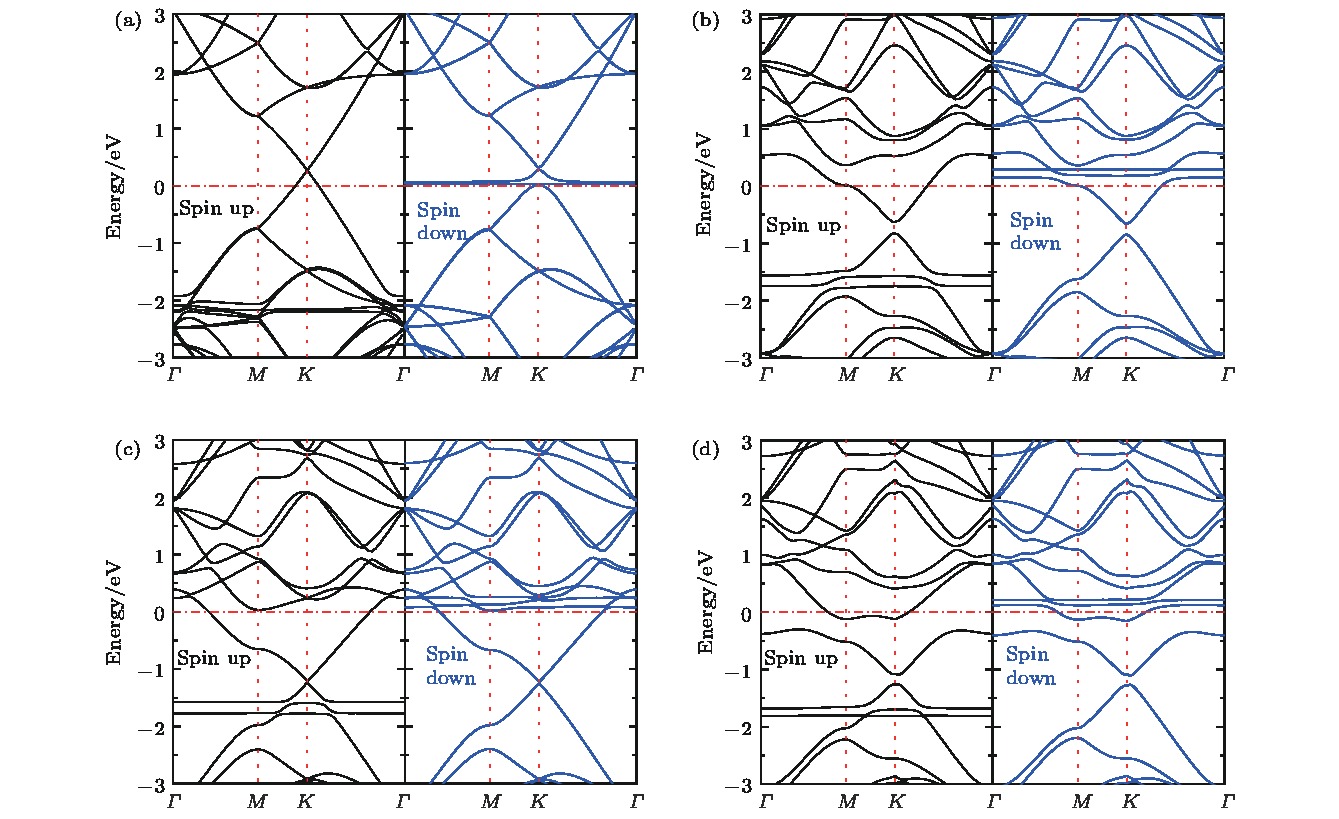
图 6 NxG-O2能带结构图 (a) G-O2; (b) N1G-O2; (c) N2G-O2; (d) N3G-O2
Fig. 6. Band structures of NxG-O2: (a) G-O2; (b) N1G-O2; (c) N2G-O2; (d) N3G-O2.
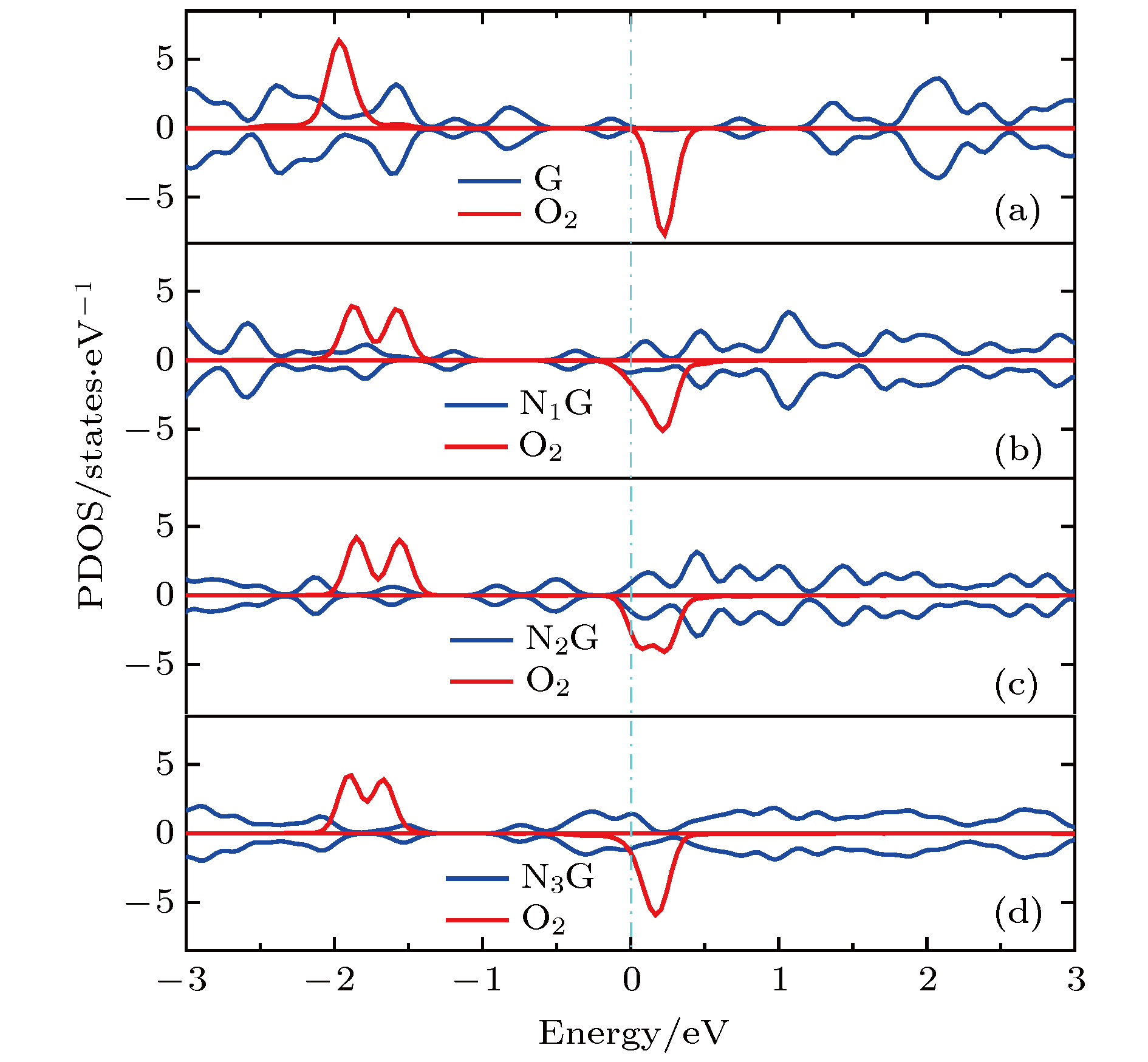
图 7 分态密度图 (a) G-O2; (b) N1G-O2; (c) N2G-O2; (d) N3G-O2
Fig. 7. Projected density of states (PDOS): (a) G-O2; (b) N1G-O2; (c) N2G-O2; (d) N3G-O2.
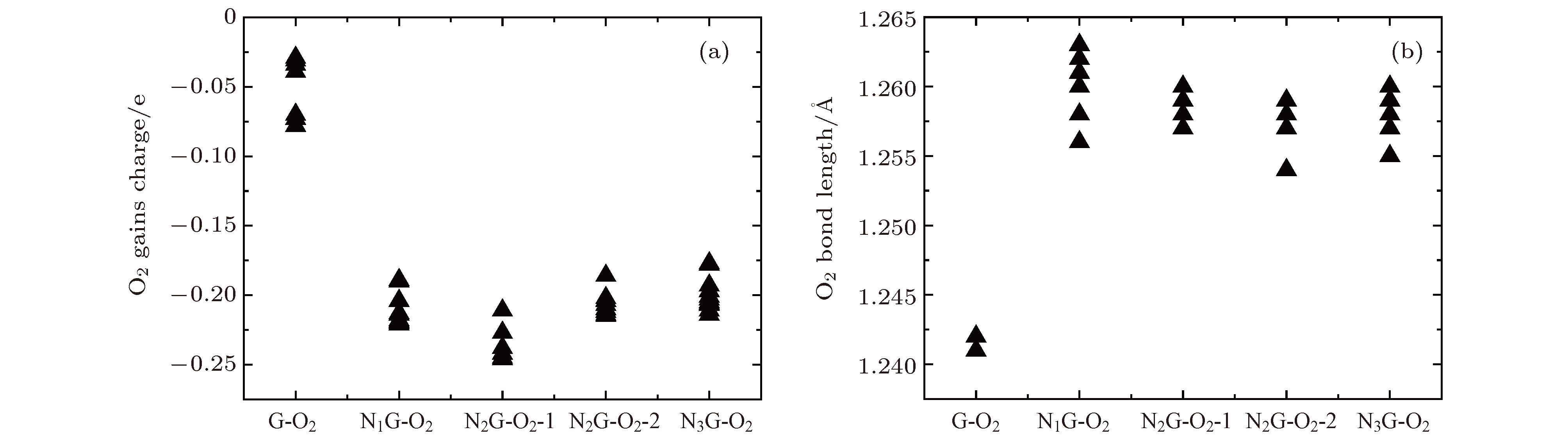
图 8 NxG-O2(x = 0, 1, 2, 3)不同异构体的氧分子获得电荷 (a)及键长(b)统计
Fig. 8. Charges (a) and O—O distances (b) of isomers of NxG-O2 (x = 0, 1, 2, 3).

图 9 GN 吸附氧分子的最稳定构型(碳原子、氮原子和氧原子分别用灰色、蓝色和红色小球表示) (a) 3 × 3 × 1 GN-O2; (b) 4 × 4 × 1 GN-O2; (c) 5 × 5 × 1 GN-O2
Fig. 9. Optimal structures of GN-O2: (a) 3 × 3 × 1 GN-O2; (b) 4 × 4 × 1 GN-O2; (c) 5 × 5 × 1 GN-O2. The C atom, N atom, and O atom are represented by grey, blue, and red spheres, respectively.
表 1 NxG (x = 0, 1, 2, 3) 吸附氧分子的相关性质
Table 1. Adsorption properties of O2 molecule on the NxG (x = 0, 1, 2, 3).
NxG-O2/at% Ead/eV dO—O/Å $Q_{{\rm O}_2} $/e QN/e N含量 O2 — 1.233 — — — G-O2 0.13 1.242 –0.04 — — N1G-O2 0.31 1.261 –0.22 –1.24 3.13 N2G-O2 0.33 1.259 –0.25 –2.39 6.25 N3G-O2 0.28 1.258 –0.21 –3.72 9.38  下载: 导出CSV
下载: 导出CSV
表 2 不同N掺杂石墨烯基底吸附氧分子的相关性质
Table 2. Adsorption properties of O2 molecule on the 3 × 3 × 1 GN-O2, 4 × 4 × 1 GN-O2, and 5 × 5 × 1 GN-O2.
NG-O2 Ead/eV dO—O/Å $ d_{{\rm O}_2\text{-}{\rm NG}}$/Å $ Q_{{\rm O}_2}$/e QN/e $d_{{\rm O}_2\text{-}{\rm N}} $/Å N含量/at% 3 × 3 × 1 0.27 1.257 2.937 –0.19 –1.23 3.011 5.56 4 × 4 × 1 0.31 1.261 2.896 –0.22 –1.24 2.969 3.13 5 × 5 × 1 0.20 1.254 2.977 –0.20 –1.14 3.056 2.00
下载: 导出CSV
-
[1] Yang Z, Zhang J, Kintner-Meyer M C W, Lu X, Choi D, Lemmon J P, Liu J 2011 Chem. Rev. 111 3577
Google Scholar
[2] Kwak W J, Kim H, Jung H G, Aurbach D, Sun Y K 2018 J. Electrochem. Soc. 165 A2274
Google Scholar
[3] Zhao N, Li C, Guo X 2014 Energy Technol. 2 317
Google Scholar
[4] Wen Z, Shen C, Lu Y 2015 ChemPlusChem 80 270
Google Scholar
[5] Lim H D, Lee B, Bae Y, Park H, Ko Y, Kim H, Kim J, Kang K 2017 Chem. Soc. Rev. 46 2873
Google Scholar
[6] Dai W, Cui X, Zhou Y, Zhao Y, Wang L, Peng L, Chen W 2019 Small Methods 3 1800358
Google Scholar
[7] Lu J, Li L, Park J B, Sun Y K, Wu F, Amine K 2014 Chem. Rev. 114 5611
Google Scholar
[8] Aurbach D, McCloskey B D, Nazar L F, Bruce P G 2016 Nat. Energy 1 1
[9] Abraham K M, Jiang Z 1996 Electrochem. Sci. Technol. 143 1
Google Scholar
[10] Ogasawara T, Débart A l, Holzapfel M, Novák P, Bruce P G 2006 J. Am. Chem. Soc. 128 1390
Google Scholar
[11] Wang Y, Lai N C, Lu Y R, Zhou Y, Dong C L, Lu Y C 2018 Joule 2 2364
Google Scholar
[12] Lu Y, Tong S, Qiu F, Jiang J, Feng N, Zhang X, He P, Zhou H 2016 J. Power Sources 329 525
Google Scholar
[13] Liu C J, Brant W R, Younesi R, Dong Y Y, Edstrom K, Gustafsson T, Zhu J F 2017 ChemSusChem 10 1592
Google Scholar
[14] Cui Y M, Wen Z Y, Liang X, Lu Y, Jin J, Wu M F, Wu X W 2012 Energy Environ. Sci. 5 7893
Google Scholar
[15] Wang J J, Li Y L, Sun X L 2013 Nano Energy 2 443
Google Scholar
[16] Ma Z, Yuan X, Li L, Ma Z F, Wilkinson D P, Zhang L, Zhang J 2015 Energy Environ. Sci. 8 2144
Google Scholar
[17] Tang Y, Qiao H, Wang H, Tao P 2013 J. Mater. Chem. A 1 12512
Google Scholar
[18] Xiao J, Mei D, Li X, Xu W, Wang D, Graff G L, Bennett W D, Nie Z, Saraf L V, Aksay I A, Liu J, Zhang J G 2011 Nano Lett. 11 5071
Google Scholar
[19] Jin L, Xu L, Morein C, Chen C, Lai M, Dharmarathna S, Dobley A, Suib S L 2010 Adv. Funct. Mater. 20 3373
Google Scholar
[20] Gao R, Shang Z, Zheng L, Wang J, Sun L, Hu Z, Liu X 2019 Inorg. Chem 58 4989
Google Scholar
[21] Park J B, Lee S H, Jung H G, Aurbach D, Sun Y K 2018 Adv. Mater. 30 1704162
Google Scholar
[22] Liu S, Wang J, Zeng J, Ou J, Li Z, Liu X, Yang S 2010 J. Power Sources 195 4628
Google Scholar
[23] Soin N, Roy S S, Lim T H, McLaughlin J A D 2011 Mater. Chem. Phys. 129 1051
Google Scholar
[24] Li Y, Wang J, Li X, Geng D, Li R, Sun X 2011 Chem. Commun. 47 9438
Google Scholar
[25] Sun B, Wang B, Su D, Xiao L, Ahn H, Wang G 2012 Carbon 50 727
Google Scholar
[26] Yang Y, Shi M, Zhou Q F, Li Y S, Fu Z W 2012 Electrochem. Commun. 20 11
Google Scholar
[27] Kim H, Lim H D, Kim J, Kang K 2014 J. Mater. Chem. A 2 33
Google Scholar
[28] Jung H G, Jeong Y S, Park J B, Sun Y K, Scrosati B, Lee Y J 2013 ACS Nano 7 3532
Google Scholar
[29] Shao Y, Zhang S, Engelhard M H, Li G, Shao G, Wang Y, Liu J, Aksay I A, Lin Y 2010 J. Mater. Chem. 20 7491
Google Scholar
[30] Li Y, Wang J, Li X, Liu J, Geng D, Yang J, Li R, Sun X 2011 Electrochem. Commun. 13 668
Google Scholar
[31] Li Y, Wang J, Li X, Geng D, Banis M N, Li R, Sun X 2012 Electrochem. Commun. 18 12
Google Scholar
[32] Higgins D, Chen Z, Lee D U, Chen Z 2013 J. Mater. Chem. A 1 2639
Google Scholar
[33] Zhao C, Yu C, Liu S, Yang J, Fan X, Huang H, Qiu J 2015 Adv. Funct. Mater. 25 6913
Google Scholar
[34] Yan H J, Xu B, Shi S Q, Ouyang C Y 2012 J. Appl. Phys. 112 104316
Google Scholar
[35] Kresse G, Hafner J 1993 Phys. Rev. B 47 558
Google Scholar
[36] Kresse G, Furthmüller J 1996 Phys. Rev. B 54 11169
Google Scholar
[37] Kresse G, Joubert D 1999 Phys. Rev. B 59 1758
[38] Blöchl P E 1994 Phys. Rev. B 50 17953
Google Scholar
[39] Perdew J P, Burke K, Ernzerhof M 1996 Phys. Rev. Lett. 77 3865
Google Scholar
[40] Perdew J P, Ernzerhof M, Burke K 1996 J. Chem. Phys. 105 9982
Google Scholar
[41] Grimme S 2006 J. Comput. Chem. 27 1787
Google Scholar
[42] Monkhorst H J, Pack J D 1976 Phys. Rev. B 13 5188
Google Scholar
[43] Pramanik A, Kang H S 2011 J. Phys. Chem. C 115 10971
Google Scholar
[44] Novoselov K S, Geim A K, Morozov S V, Jiang D, Zhang Y, Dubonos S V, Grigorieva I V, Firsov A A 2004 Science 306 666
Google Scholar
[45] Avouris P, Chen Z, Perebeinos V 2007 Nature Nanotech. 2 605
Google Scholar
[46] Tang W, Sanville E, Henkelman G 2009 J. Phys.: Condens. Matter 21 084204
Google Scholar
-
128801-Suppl.pdf
128801补充材料图S1—图S7






 下载:
下载:
计量
- 文章访问数: 25459
- PDF下载量: 174
- 被引次数: 0
