










-
一种理想的Li离子电池负极材料需要具有较高的储Li容量和较低的体积膨胀比. 本文应用密度泛函理论研究了二维多孔石墨炔在B, N原子掺杂调控后作为Li离子电池负极材料时的储Li性能. 计算结果表明, B在石墨炔结构中的掺杂可以增强Li与石墨炔之间的吸附作用, 储Li容量可以增加到2061.62 mAh/g, 与未掺杂单层石墨炔相比增加了2.77倍. 同时, B掺杂降低了Li在垂直于石墨炔平面方向上的扩散能垒, 而面内扩散能垒提高了0.1 eV. N掺杂降低了Li与石墨炔之间的相互作用, 但增加了Li的稳定位点, 储Li容量增加到了1652.12 mAh/g, 同时, Li在石墨炔上的扩散性能大大提高, 在平面内扩散能垒降至0.37 eV, 因此N掺杂石墨炔的充放电性能得到较好提升. 因此, B, N掺杂可从不同方面提升石墨炔作为Li电池负极材料时的储Li性能. 该研究可以为开发良好的储Li负极材料提供一个良好的研究思路, 为实验工作者提供理论依据.As the economy grows and the environment deteriorates, the renewable energy is urgently needed. The advanced energy storage technology in electronic equipment, electric vehicle, smart grid, etc. becomes more significant. For example, the rechargeable batteries, hydrogen storage media, supercapacitors, the new energy storage devices have received much attention today. The anodes of the lithium ion battery (LIB), as the main body of charging and discharging, should be most important. The ideal anode material for LIBs is required to possess a higher Li capacity and a lower volume expansion. Good reversibility and high Li capacity are balanced necessarily in the electrode material. The poor cycling performance of LIB is usually due to the severe volume expansion of anode in lithiation/delithiation process. In this paper, the Li storage performance of B and N doped graphyne is explored by using the density functional theory method. The Perdew-Burke-Ernzerhof functional of the generalized gradient approximation is chosen. The calculations indicate that the doping of B atoms can enhance the adsorption strength between the Li atom and the graphyne, which can greatly increase the Li storage capacity. The Li storage capacity of B doped graphyne can reach as high as 2061.62 mAh/g, which is 2.77 times that of pristine monolayer graphyne. Meanwhile, the B doping reduces the out-plane diffusion energy barrier of Li, but increases the in-plane diffusion energy barrier slightly by 0.1 eV. On the other hand, the doping of N atoms reduces the interaction between Li and graphyne, however, the Li capacity also increases to 1652.12 mAh/g because the number of the available Li adsorption sites increases. Moreover, the doping of N atoms greatly improves the diffusion performance of Li on graphyne. The in-plane diffusion energy barrier drops to 0.37 eV, and thus the charge-discharge performance of the N doping graphyne is well improved. Therefore, the doping of B and N atoms can remarkably improve the performance of graphyne as the LIB anodes. The remarkable performance of B and N doped graphdiyne shows that it will become a promising LIB anode in the future. The present research can provide a good theoretical basis and thus conduce to guiding the developing of good Li storage materials, and can also supply strong background for experimental researches.
-
Keywords:
- graphyne /
- B doped graphyne /
- N doped graphyne /
- Li storage /
- density functional theory
[1] Bruce P G, Freunberger S A, Hardwick andv L J, M Tarascon J 2012 Nat. Mater. 11 19
 Google Scholar
Google Scholar
[2] Zheng G, Lee S W, Liang Z, Lee H W, Yan K, Yao H, Wang H, Li W, Chu S, Cui Y 2014 Nat. Nanotechnol. 9 618
Google Scholar
[3] Cheng F Y, Liang J, Tao Z L, Chen J 2011 Adv. Mater. 23 1695
Google Scholar
[4] Osumi S, Saito S, Dou C, Matsuo K, Kume K, Yoshikawa H, Awaga K, Yamaguchi S 2016 Chem. Sci. 7 219
Google Scholar
[5] Binitha G, Ashish A G, Ramasubramonian D, Manikandan P, Shaijumon M M 2016 Adv. Mater. Interfaces 3 1500419
Google Scholar
[6] Liang X, Hart C, Pang Q, Garsuch A, Weiss T, Nazar L F 2015 Nat. Commun. 6 5682
Google Scholar
[7] Liu R Z, Zhao Y H, Chu T S 2015 Chem. Commun. 51 2429
Google Scholar
[8] Zhu Y, Murali S, Stoller M D, Ganesh K J, Cai W, Ferreira P J, Pirkle A, Wallace R M, Cychosz K A, Thommes M, D Su, Stach E A, Ruoff R S 2011 Science 332 1537
Google Scholar
[9] Hankel M, Searles D J 2016 Phys. Chem. Chem. Phys. 18 14205
Google Scholar
[10] Hwang H J, Koo J, Park M, Park N, Kwon Y, Lee H 2013 J. Phys. Chem. C 117 6919
Google Scholar
[11] Eftekhari A 2017 Ener. Storage Mater. 7 157
Google Scholar
[12] Jang B, Koo J, Park M, Lee H, Nam J, Kwon Y, Lee H 2013 Appl. Phys. Lett. 103 263904
Google Scholar
[13] Zhang W J 2011 J. Power. Sources 196 13
Google Scholar
[14] Wu H, Cui Y 2012 Nano Today 7 414
Google Scholar
[15] Paraknowitsch J P, Thomas A 2013 Ener. Environ. Sci. 6 2839
Google Scholar
[16] Zhu G, Lü K, Sun Q, Kawazoe Y, Jena P 2014 Comp. Mater. Sci. 81 275
Google Scholar
[17] Wang X, Weng Q, Liu X, Wang X, Tang D M, Tian W, Zhang C, Yi W, Liu D, Bando Y, Golberg D 2014 Nano Lett. 14 1164
[18] Ma C, Shao X, Cao D 2012 J. Mater. Chem. 22 8911
Google Scholar
[19] Veith G M, Baggetto L, Adamczyk L, Guo A B, Brown S S, Sun X G, Albert A A, Humble J R, Barnes C E, Bojdys M J, Dai S, Dudney N J 2013 Chem. Mater. 25 503
Google Scholar
[20] Tian L L, W ei, X Y, Zhuang Q C, Jiang C H, Wu C, Ma G Y, Zhao X, Zong Z M, Sun S G 2014 Nanoscale 6 6075
[21] Zhang S, Du H, He J, Huang C, Liu H, Cui G, Li Y 2016 ACS Appl. Mater. Inter. 8 8467
Google Scholar
[22] Yang L, Jiang S, Zhao Y, Zhu L, Chen S, Wang X, Wu Q, Ma J, Ma Y, Hu Z 2011 Angew. Chem. Int. Ed. 50 7132
Google Scholar
[23] Sheng Z H, Gao H L, Bao W J, Wang F B, Xia X H 2012 J. Mater. Chem. 22 390
Google Scholar
[24] Luo G, Zhao J, Wang B 2013 Compu. Mater. Sci. 68 212
Google Scholar
[25] Baughman R H, Eckhardt H, Kertesz M 1987 J. Chem. Phys. 87 6687
Google Scholar
[26] Li Q, Li Y, Chen Y, Wu L, Yang C, Cui X 2018 Carbon 136 248
Google Scholar
[27] Bhattacharya B, Sarkar U 2016 J. Phys. Chem. C 120 26793
[28] Jafari M, Asadpour M, Majelan N A, Faghihnasiri M 2014 Comput. Mater. Sci. 82 391
Google Scholar
[29] Ruiz-Puigdollers A, Gamallo P 2017 Carbon 114 301
Google Scholar
[30] Becke A D 1988 Phys. Rev. A 38 3098
Google Scholar
[31] Delley B 1990 J. Chem. Phys. 92 508
Google Scholar
[32] Delley B 1998 Int. J. Quant. Chem. 69 423
Google Scholar
[33] Perdew J P, Chevary J A, Vosko S H, Jackson K A, Pederson M R, Singh D J 1992 Phys. Rev. B 46 6671
Google Scholar
[34] Perdew J P, Burke K, Ernzerhof M 1996 Phys. Rev. Lett. 77 3865
Google Scholar
[35] Grimme S 2006 J. Comput. Chem. 27 1787
Google Scholar
[36] Olmstead M M, Power P P, Weese K J, Doedens R J 1987 J. Am. Chem. Soc. 109 2541
Google Scholar
[37] Majidi R 2013 Nano 8 1350060
[38] Merritt L L, Lanterman E 1952 Acta Crystallogr. 5 811
Google Scholar
[39] Deng X Z, Zhao Q Q, Zhao Y Q, Cai M Q 2019 Curr. Appl. Phys. 19 279
Google Scholar
[40] Yu Z L, Ma Q R, Liu B, Zhao Y Q, Wang L Z, Zhou H, Cai M Q 2017 J. Phys. D: Appl. Phys. 50 465101
Google Scholar
[41] Zhao Y Q, Wang X, Liu B, Yu Z L, He P B, Wan Q, Yu H L 2018 Org. Electron. 53 50
Google Scholar
[42] Zhao Y Q, Ma Q R, Liu B, Yu Z L, Yang J, Cai M Q 2018 Nanoscale 10 8677
Google Scholar
[43] Guo Y, Cao J, Bo X, Xia Y, Jiang Y, Liu Z 2013 Compu. Mater. Sci. 68 61
Google Scholar
[44] Jiang X, Arhammar C, Liu P, Zhao J, Ahuja R 2013 Sci. Rep. 3 1877
Google Scholar
[45] Kittel C 1996 Introduction to Solid State Physics (7th ed.) (Singapore: Wiley) pp356−358
[46] Zhang Q, Tang C, Zhu W, Cheng C 2018 J. Phys. Chem. C 122 22838
Google Scholar
[47] Zheng F, Yang Y, Chen Q 2014 Nat. Commun. 5 5261
Google Scholar
[48] Mortazavi B, Shahrokhi M, Zhuang X, Rabczuk T 2018 J. Mater. Chem. A 6 11022
Google Scholar
[49] Eftekhari A, Molaei F 2015 J. Power Sources 274 1306
Google Scholar
[50] Eftekhari A, Molaei F 2015 J. Power Sources 274 1315
Google Scholar
[51] Halgren T A, Lipscomb W N 1977 Chem. Phys. Lett. 49 225
Google Scholar
[52] Henkelman G 2000 J. Chem. Phys. 113 9978
Google Scholar
[53] Sun C, Searles D J 2012 J. Phys. Chem. C 116 26222
Google Scholar
[54] Chan K T, Neaton J B, Cohen M L 2008 Phys. Rev. B 77 235430
Google Scholar
[55] Toyoura K, Koyama Y, Kuwabara A, Oba F, Tanaka I 2008 Phys. Rev. B 78 214303
Google Scholar
[56] Valencia F, Romero A H, Ancilotto F, Silvestrelli P L 2006 J. Phys. Chem. B 110 14832
Google Scholar
-
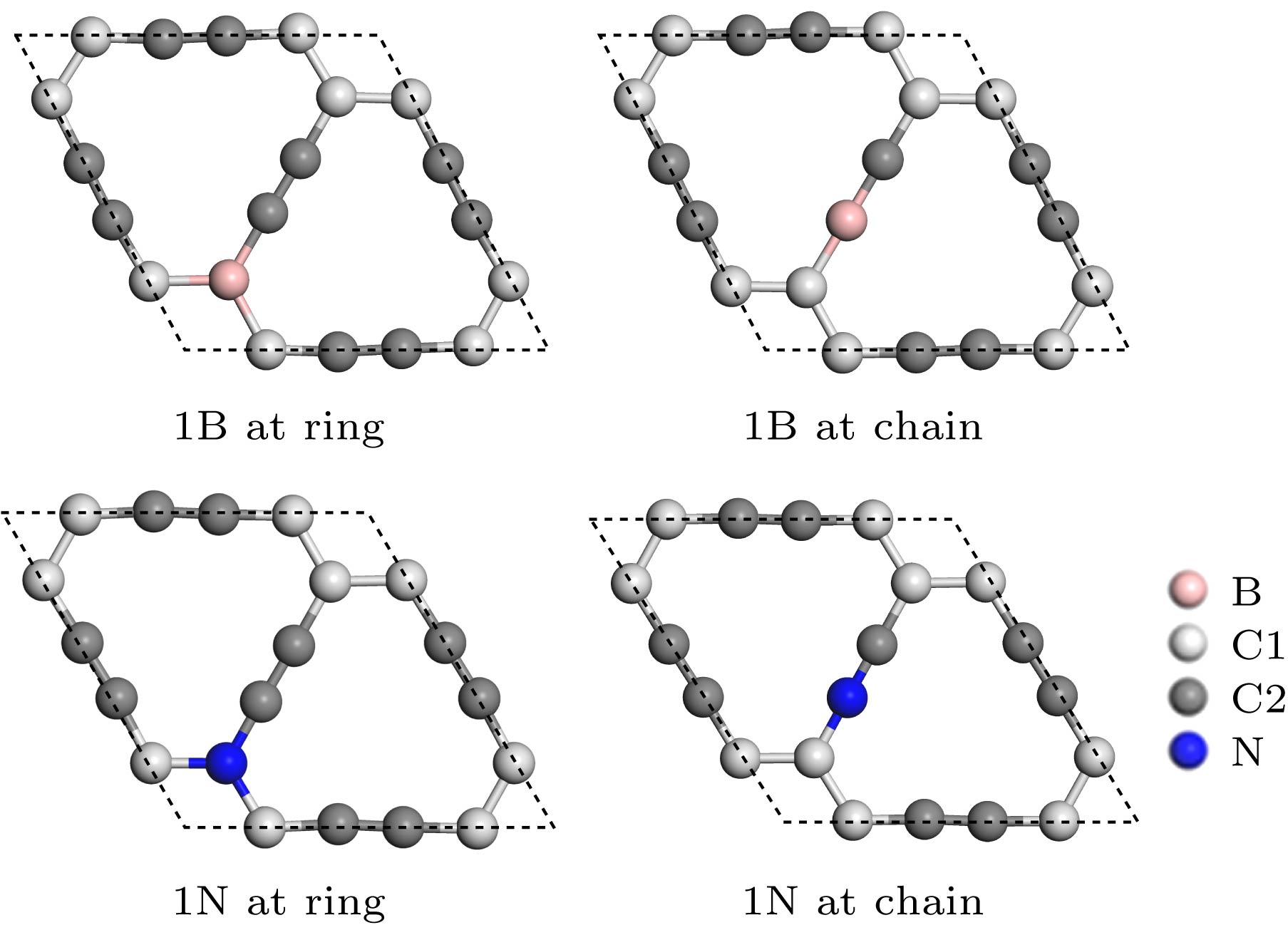
图 1 2 × 2 × 1的石墨炔晶胞中单个B, N的两种掺杂位点, 分别为环掺杂和链掺杂
Fig. 1. Two doping sites of single B and N in the 2 × 2 × 1 supercell of graphyne. They are ring doping and chain doping respectively.
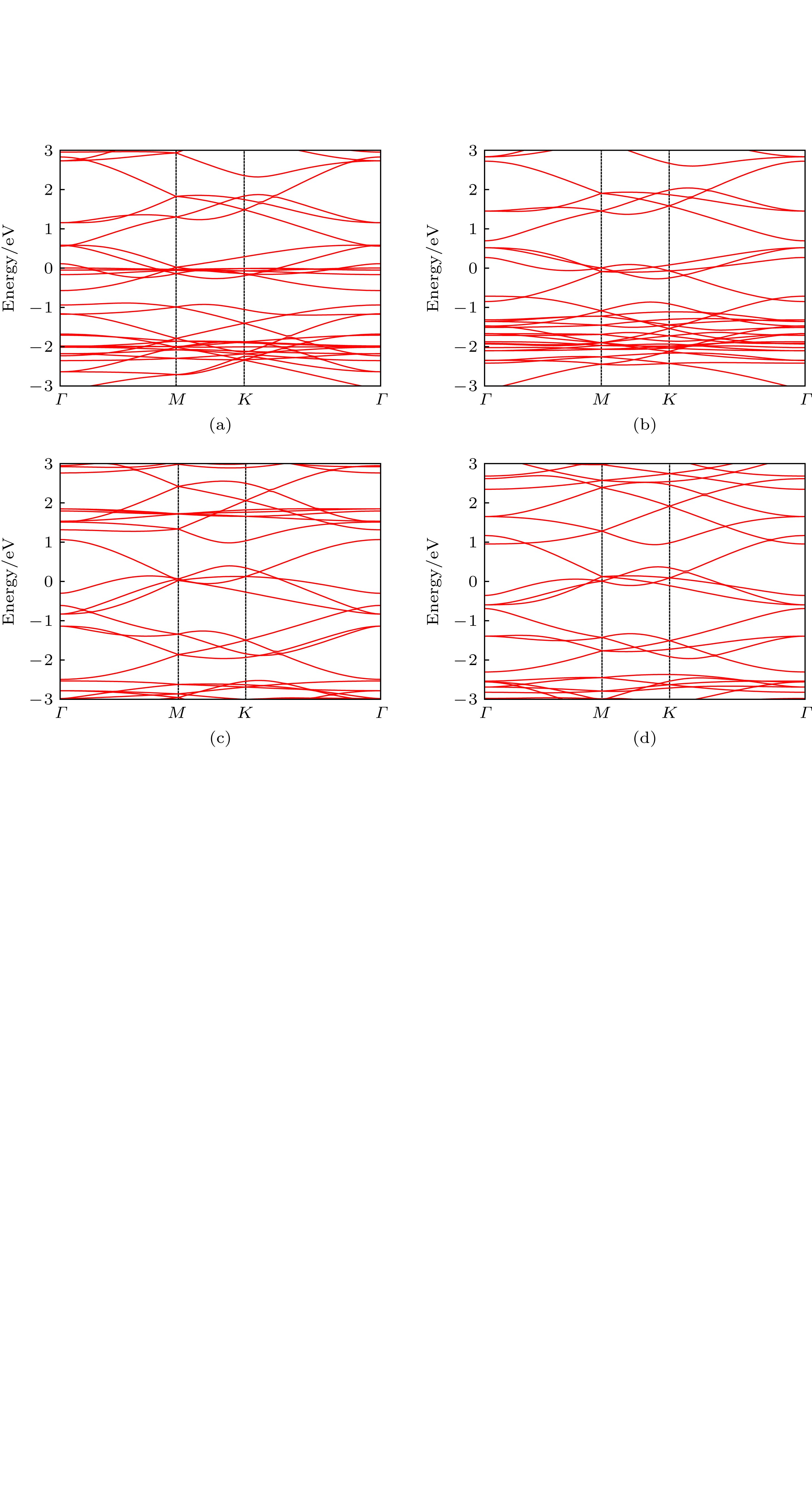
图 2 四种结构的能带图 (a) B进行环掺杂; (b) B进行链掺杂; (c) N进行环掺杂; (d) N进行链掺杂
Fig. 2. Energy band of four structures: (a) B-ring doping; (b) B-chain doping; (c) N-ring doping; (d) N-chain doping.
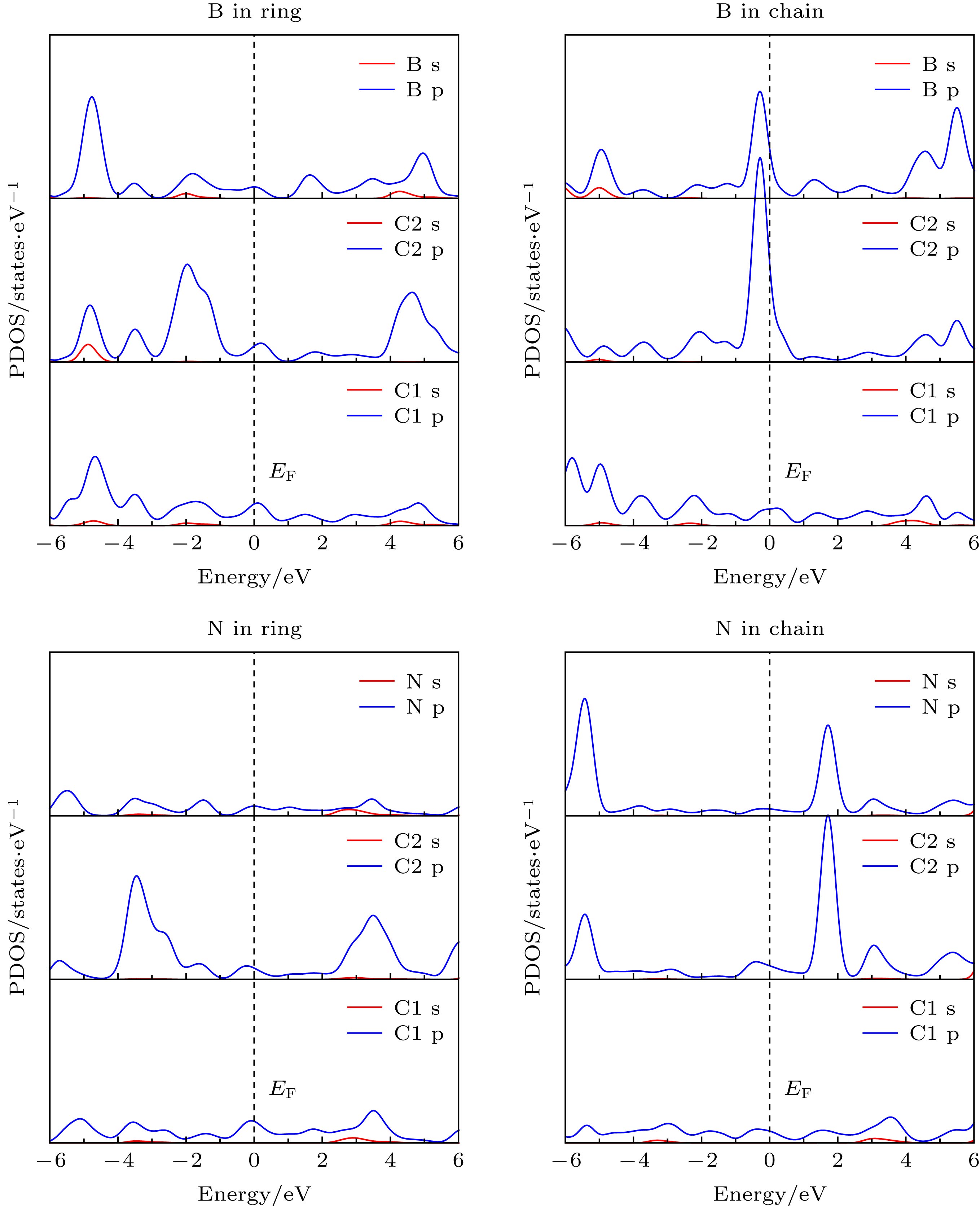
图 3 不同掺杂位点B, N与相邻C1, C2原子的PDOS图
Fig. 3. The PDOS of B, N and neighboring C1, C2 atoms with different doping sites.

图 4 B, N掺杂石墨炔静电势的平视图与侧视图(静电势范围为1.0−–1.0 Ha·e–1)
Fig. 4. Flat view and side view of the electrostatic potential of B, N doped graphyne. The range of electrostatic potential is 1.0−–1.0 Ha·e–1.

图 5 一个Li位于B掺杂的石墨炔(a) H和(c) h位点时的差分电荷密度图; 一个Li位于N掺杂石墨炔(b) H和(d) h位点的差分电荷密度图; 其中差分电荷密度范围为–0.01−0.005 e/Å3, 红色表示电子积聚, 蓝色表示电子缺失
Fig. 5. Differential charge densities: One Li at (a) H and (c) h sites of the B-doped graphyne; one Li at (b) H and (d) h sites of the N doped graphyne. The range is –0.01− 0.005 e/Å3, the red area stands for electron accumulation, and the blue area stands for electron deletion.
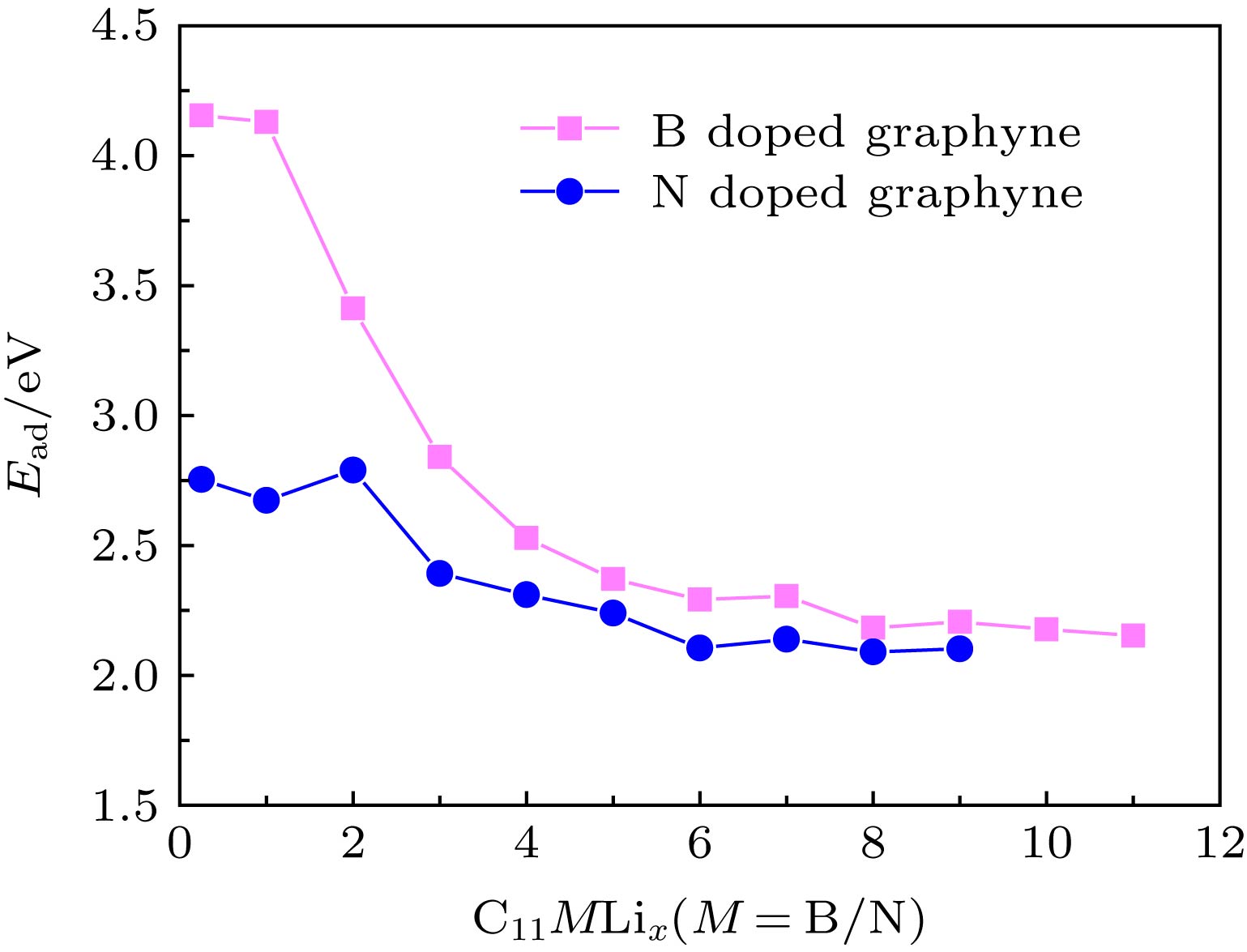
图 6 多个Li在B, N掺杂墨炔上的平均吸附能随储Li数量的变化
Fig. 6. The Ead curves of multiple Li adsorbed on B, N doped graphyne.

图 7 (a) B掺杂石墨炔最大Li结构的俯视图和侧视图; (b) N掺杂石墨炔最大储Li结构的俯视图和侧视图
Fig. 7. (a) Top and side view of the maximum Li adsorbed with B graphyne; (b) top and side view of the maximum storage Li adsorbed N doped graphyne.
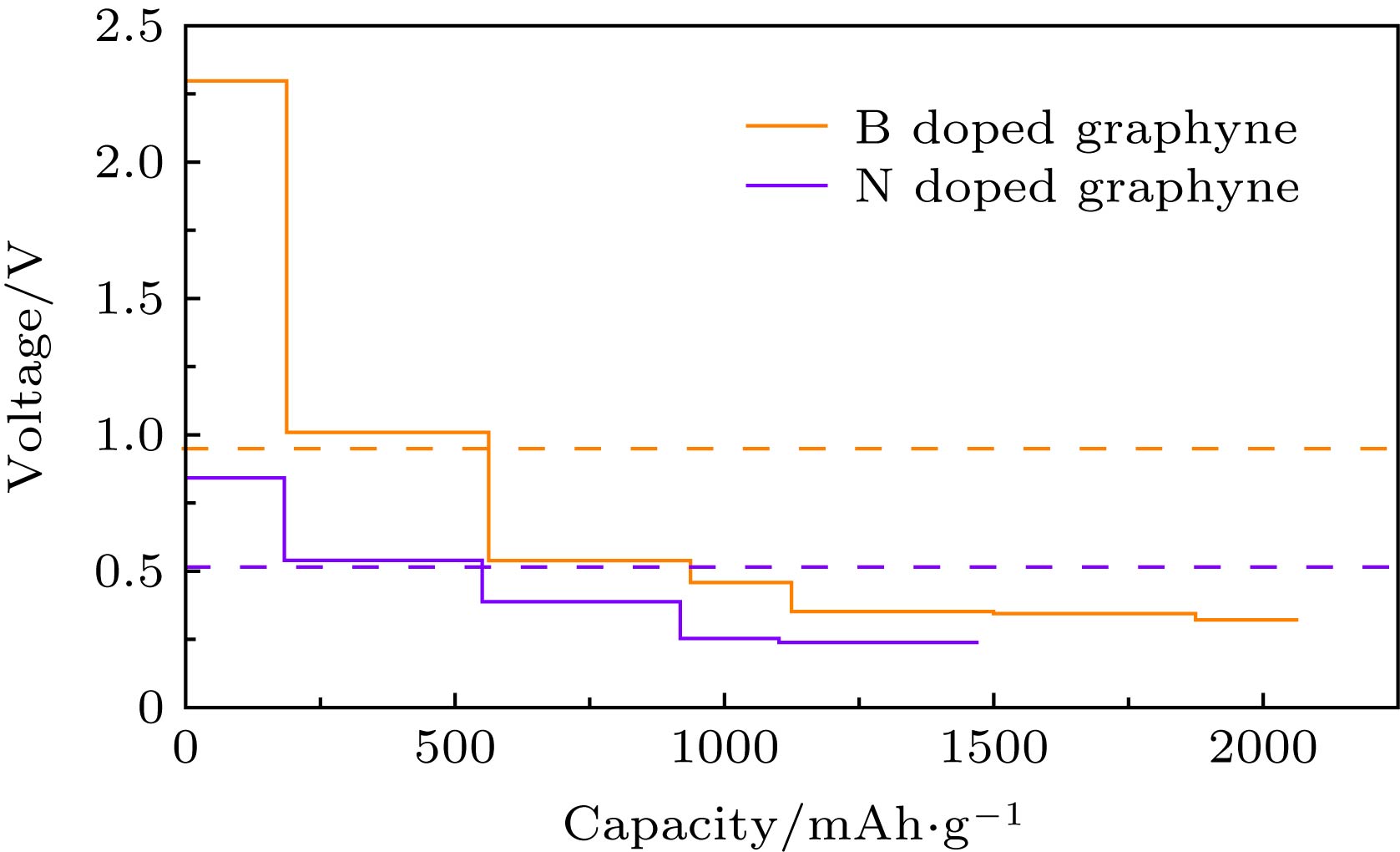
图 8 B, N掺杂石墨炔的开路电压随储Li容量的变化, 其中橙色划线表示B掺杂石墨炔的平均开路电压, 紫色划线表示N掺杂石墨炔的平均开路电压
Fig. 8. Change curves of the open circuit voltage with the storage Li capacity for B, N doped graphyne. The orange dash line represents the average open circuit voltage of B doped graphyne, and the purple dash line represents the average open circuit voltage of N doped graphyne.
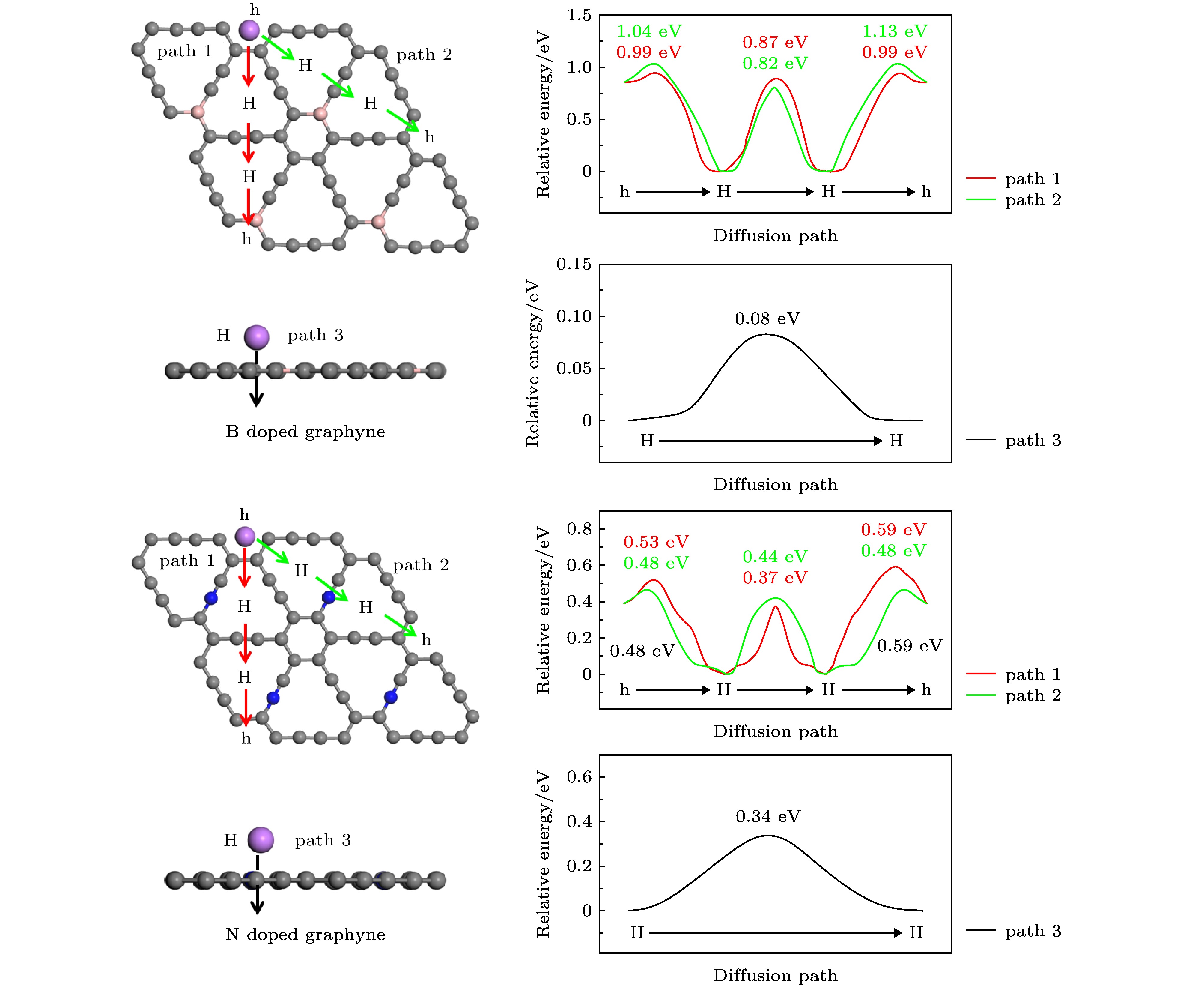
图 9 Li在B, N掺杂石墨炔上的扩散路径和对应的能量曲线图, 图中红色曲线对应path 1上的扩散能垒; 绿色曲线对应path 2上的扩散能垒; 黑色曲线对应path 3上的扩散能垒
Fig. 9. Diffusion paths of Li on B, N doped graphyne and the corresponding energy curves. The red, green, black curves in the panels corresponds to the diffusion energy barrier on path 1, 2, 3, respectively.
表 1 B, N掺杂的石墨炔的晶格常数、键长、Mulliken电荷及Eb
Table 1. Lattice constant, bond length, Mulliken charge and Eb of B, N doped graphyne.
1 B at ring 1 B at chain 1 N at ring 1 N at chain Lattice/Å 6.98 6.92 6.86 6.90 Bond length/Å B/N-C1 1.54 1.50 1.42 1.34 B/N-C2 1.50 1.36 1.34 1.18 Charge of B/N/e 0.143 0.016 –0.247 0.226 Eb/eV 7.17 7.09 6.99 7.08  下载: 导出CSV
下载: 导出CSV
-
[1] Bruce P G, Freunberger S A, Hardwick andv L J, M Tarascon J 2012 Nat. Mater. 11 19
Google Scholar
[2] Zheng G, Lee S W, Liang Z, Lee H W, Yan K, Yao H, Wang H, Li W, Chu S, Cui Y 2014 Nat. Nanotechnol. 9 618
Google Scholar
[3] Cheng F Y, Liang J, Tao Z L, Chen J 2011 Adv. Mater. 23 1695
Google Scholar
[4] Osumi S, Saito S, Dou C, Matsuo K, Kume K, Yoshikawa H, Awaga K, Yamaguchi S 2016 Chem. Sci. 7 219
Google Scholar
[5] Binitha G, Ashish A G, Ramasubramonian D, Manikandan P, Shaijumon M M 2016 Adv. Mater. Interfaces 3 1500419
Google Scholar
[6] Liang X, Hart C, Pang Q, Garsuch A, Weiss T, Nazar L F 2015 Nat. Commun. 6 5682
Google Scholar
[7] Liu R Z, Zhao Y H, Chu T S 2015 Chem. Commun. 51 2429
Google Scholar
[8] Zhu Y, Murali S, Stoller M D, Ganesh K J, Cai W, Ferreira P J, Pirkle A, Wallace R M, Cychosz K A, Thommes M, D Su, Stach E A, Ruoff R S 2011 Science 332 1537
Google Scholar
[9] Hankel M, Searles D J 2016 Phys. Chem. Chem. Phys. 18 14205
Google Scholar
[10] Hwang H J, Koo J, Park M, Park N, Kwon Y, Lee H 2013 J. Phys. Chem. C 117 6919
Google Scholar
[11] Eftekhari A 2017 Ener. Storage Mater. 7 157
Google Scholar
[12] Jang B, Koo J, Park M, Lee H, Nam J, Kwon Y, Lee H 2013 Appl. Phys. Lett. 103 263904
Google Scholar
[13] Zhang W J 2011 J. Power. Sources 196 13
Google Scholar
[14] Wu H, Cui Y 2012 Nano Today 7 414
Google Scholar
[15] Paraknowitsch J P, Thomas A 2013 Ener. Environ. Sci. 6 2839
Google Scholar
[16] Zhu G, Lü K, Sun Q, Kawazoe Y, Jena P 2014 Comp. Mater. Sci. 81 275
Google Scholar
[17] Wang X, Weng Q, Liu X, Wang X, Tang D M, Tian W, Zhang C, Yi W, Liu D, Bando Y, Golberg D 2014 Nano Lett. 14 1164
[18] Ma C, Shao X, Cao D 2012 J. Mater. Chem. 22 8911
Google Scholar
[19] Veith G M, Baggetto L, Adamczyk L, Guo A B, Brown S S, Sun X G, Albert A A, Humble J R, Barnes C E, Bojdys M J, Dai S, Dudney N J 2013 Chem. Mater. 25 503
Google Scholar
[20] Tian L L, W ei, X Y, Zhuang Q C, Jiang C H, Wu C, Ma G Y, Zhao X, Zong Z M, Sun S G 2014 Nanoscale 6 6075
[21] Zhang S, Du H, He J, Huang C, Liu H, Cui G, Li Y 2016 ACS Appl. Mater. Inter. 8 8467
Google Scholar
[22] Yang L, Jiang S, Zhao Y, Zhu L, Chen S, Wang X, Wu Q, Ma J, Ma Y, Hu Z 2011 Angew. Chem. Int. Ed. 50 7132
Google Scholar
[23] Sheng Z H, Gao H L, Bao W J, Wang F B, Xia X H 2012 J. Mater. Chem. 22 390
Google Scholar
[24] Luo G, Zhao J, Wang B 2013 Compu. Mater. Sci. 68 212
Google Scholar
[25] Baughman R H, Eckhardt H, Kertesz M 1987 J. Chem. Phys. 87 6687
Google Scholar
[26] Li Q, Li Y, Chen Y, Wu L, Yang C, Cui X 2018 Carbon 136 248
Google Scholar
[27] Bhattacharya B, Sarkar U 2016 J. Phys. Chem. C 120 26793
[28] Jafari M, Asadpour M, Majelan N A, Faghihnasiri M 2014 Comput. Mater. Sci. 82 391
Google Scholar
[29] Ruiz-Puigdollers A, Gamallo P 2017 Carbon 114 301
Google Scholar
[30] Becke A D 1988 Phys. Rev. A 38 3098
Google Scholar
[31] Delley B 1990 J. Chem. Phys. 92 508
Google Scholar
[32] Delley B 1998 Int. J. Quant. Chem. 69 423
Google Scholar
[33] Perdew J P, Chevary J A, Vosko S H, Jackson K A, Pederson M R, Singh D J 1992 Phys. Rev. B 46 6671
Google Scholar
[34] Perdew J P, Burke K, Ernzerhof M 1996 Phys. Rev. Lett. 77 3865
Google Scholar
[35] Grimme S 2006 J. Comput. Chem. 27 1787
Google Scholar
[36] Olmstead M M, Power P P, Weese K J, Doedens R J 1987 J. Am. Chem. Soc. 109 2541
Google Scholar
[37] Majidi R 2013 Nano 8 1350060
[38] Merritt L L, Lanterman E 1952 Acta Crystallogr. 5 811
Google Scholar
[39] Deng X Z, Zhao Q Q, Zhao Y Q, Cai M Q 2019 Curr. Appl. Phys. 19 279
Google Scholar
[40] Yu Z L, Ma Q R, Liu B, Zhao Y Q, Wang L Z, Zhou H, Cai M Q 2017 J. Phys. D: Appl. Phys. 50 465101
Google Scholar
[41] Zhao Y Q, Wang X, Liu B, Yu Z L, He P B, Wan Q, Yu H L 2018 Org. Electron. 53 50
Google Scholar
[42] Zhao Y Q, Ma Q R, Liu B, Yu Z L, Yang J, Cai M Q 2018 Nanoscale 10 8677
Google Scholar
[43] Guo Y, Cao J, Bo X, Xia Y, Jiang Y, Liu Z 2013 Compu. Mater. Sci. 68 61
Google Scholar
[44] Jiang X, Arhammar C, Liu P, Zhao J, Ahuja R 2013 Sci. Rep. 3 1877
Google Scholar
[45] Kittel C 1996 Introduction to Solid State Physics (7th ed.) (Singapore: Wiley) pp356−358
[46] Zhang Q, Tang C, Zhu W, Cheng C 2018 J. Phys. Chem. C 122 22838
Google Scholar
[47] Zheng F, Yang Y, Chen Q 2014 Nat. Commun. 5 5261
Google Scholar
[48] Mortazavi B, Shahrokhi M, Zhuang X, Rabczuk T 2018 J. Mater. Chem. A 6 11022
Google Scholar
[49] Eftekhari A, Molaei F 2015 J. Power Sources 274 1306
Google Scholar
[50] Eftekhari A, Molaei F 2015 J. Power Sources 274 1315
Google Scholar
[51] Halgren T A, Lipscomb W N 1977 Chem. Phys. Lett. 49 225
Google Scholar
[52] Henkelman G 2000 J. Chem. Phys. 113 9978
Google Scholar
[53] Sun C, Searles D J 2012 J. Phys. Chem. C 116 26222
Google Scholar
[54] Chan K T, Neaton J B, Cohen M L 2008 Phys. Rev. B 77 235430
Google Scholar
[55] Toyoura K, Koyama Y, Kuwabara A, Oba F, Tanaka I 2008 Phys. Rev. B 78 214303
Google Scholar
[56] Valencia F, Romero A H, Ancilotto F, Silvestrelli P L 2006 J. Phys. Chem. B 110 14832
Google Scholar
-
[1] 董肖. P掺杂LiNH2团簇与LiH反应机理的密度泛函理论研究及一种新储放氢机制. 物理学报, 2023, 72(15): 153101. doi: 10.7498/aps.72.20230374 [2] 周树仁, 张红, 莫慧兰, 刘浩文, 熊元强, 李泓霖, 孔春阳, 叶利娟, 李万俊. N掺杂对 ${\boldsymbol\beta} $ -Ga2O3薄膜日盲紫外探测器性能的影响. 物理学报, 2021, 70(17): 178503. doi: 10.7498/aps.70.20210434[3] 罗强, 杨恒, 郭平, 赵建飞. N型甲烷水合物结构和电子性质的密度泛函理论计算. 物理学报, 2019, 68(16): 169101. doi: 10.7498/aps.68.20182230 [4] 张陈俊, 王养丽, 陈朝康. InCn+(n=110)团簇的密度泛函理论研究. 物理学报, 2018, 67(11): 113101. doi: 10.7498/aps.67.20172662 [5] 迟宝倩, 刘轶, 徐京城, 秦绪明, 孙辰, 白晟灏, 刘一璠, 赵新洛, 李小武. 石墨炔衍生物结构稳定性及电子结构的密度泛函理论研究. 物理学报, 2016, 65(13): 133101. doi: 10.7498/aps.65.133101 [6] 杨振清, 白晓慧, 邵长金. (TiO2)12量子环及过渡金属化合物掺杂对其电子性质影响的密度泛函理论研究. 物理学报, 2015, 64(7): 077102. doi: 10.7498/aps.64.077102 [7] 杨光敏, 徐强, 李冰, 张汉壮, 贺小光. 不同N掺杂构型石墨烯的量子电容研究. 物理学报, 2015, 64(12): 127301. doi: 10.7498/aps.64.127301 [8] 温俊青, 张建民, 姚攀, 周红, 王俊斐. PdnAl(n=18)二元团簇的密度泛函理论研究. 物理学报, 2014, 63(11): 113101. doi: 10.7498/aps.63.113101 [9] 温俊青, 夏涛, 王俊斐. PtnAl (n=18)小团簇的密度泛函理论研究. 物理学报, 2014, 63(2): 023103. doi: 10.7498/aps.63.023103 [10] 解晓东, 郝玉英, 章日光, 王宝俊. Li掺杂8-羟基喹啉铝的密度泛函理论研究. 物理学报, 2012, 61(12): 127201. doi: 10.7498/aps.61.127201 [11] 唐冬华, 薛林, 孙立忠, 钟建新. B在Hg0.75Cd0.25Te中掺杂效应的第一性原理研究. 物理学报, 2012, 61(2): 027102. doi: 10.7498/aps.61.027102 [12] 张玲, 何智兵, 廖国, 谌家军, 许华, 李俊. B掺杂对Ti薄膜结构与性能的影响. 物理学报, 2012, 61(18): 186803. doi: 10.7498/aps.61.186803 [13] 张致龙, 陈玉红, 任宝兴, 张材荣, 杜瑞, 王伟超. (HMgN3)n(n=15)团簇结构与性质的密度泛函理论研究. 物理学报, 2011, 60(12): 123601. doi: 10.7498/aps.60.123601 [14] 林琦, 陈余行, 吴建宝, 孔宗敏. N掺杂对zigzag型石墨烯纳米带的能带结构和输运性质的影响. 物理学报, 2011, 60(9): 097103. doi: 10.7498/aps.60.097103 [15] 李喜波, 王红艳, 罗江山, 吴卫东, 唐永建. 密度泛函理论研究ScnO(n=1—9)团簇的结构、稳定性与电子性质. 物理学报, 2009, 58(9): 6134-6140. doi: 10.7498/aps.58.6134 [16] 陈玉红, 康 龙, 张材荣, 罗永春, 马 军. [Mg(NH2)2]n(n=1—5)团簇的密度泛函理论研究. 物理学报, 2008, 57(8): 4866-4874. doi: 10.7498/aps.57.4866 [17] 徐 凌, 唐超群, 戴 磊, 唐代海, 马新国. N掺杂锐钛矿TiO2电子结构的第一性原理研究. 物理学报, 2007, 56(2): 1048-1053. doi: 10.7498/aps.56.1048 [18] 彭丽萍, 徐 凌, 尹建武. N掺杂锐钛矿TiO2光学性能的第一性原理研究. 物理学报, 2007, 56(3): 1585-1589. doi: 10.7498/aps.56.1585 [19] 陈玉红, 张材荣, 马 军. MgmBn(m=1,2;n=1—4)团簇结构与性质的密度泛函理论研究. 物理学报, 2006, 55(1): 171-178. doi: 10.7498/aps.55.171 [20] 赖云锋, 冯 洁, 乔保卫, 凌 云, 林殷茵, 汤庭鳌, 蔡炳初, 陈邦明. 氮掺杂Ge2Sb2Te5相变存储器的多态存储功能. 物理学报, 2006, 55(8): 4347-4352. doi: 10.7498/aps.55.4347

 下载:
下载:

计量
- 文章访问数: 13177
- PDF下载量: 215
- 被引次数: 0
