
















-
基于一个最新的CH2
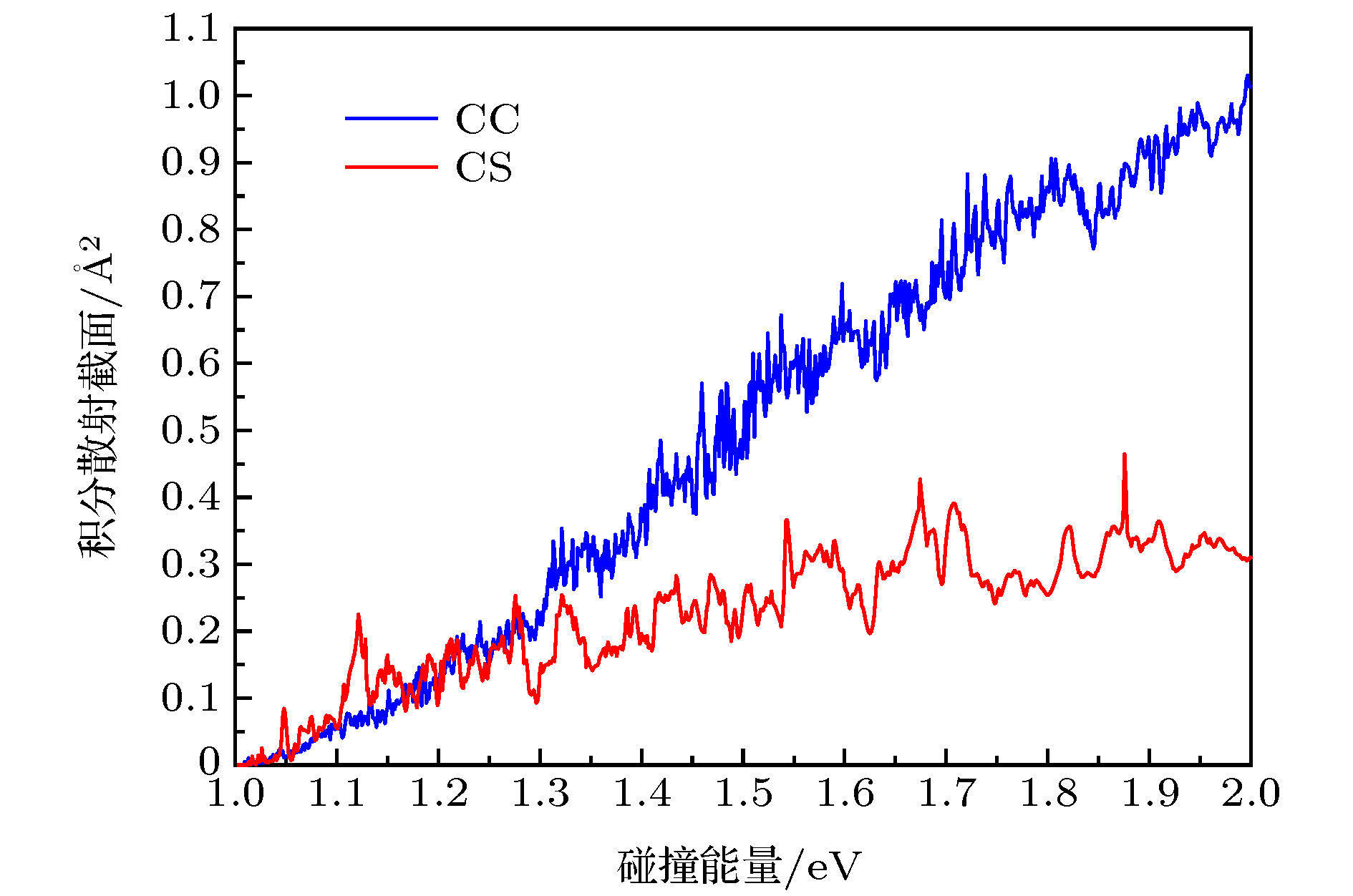
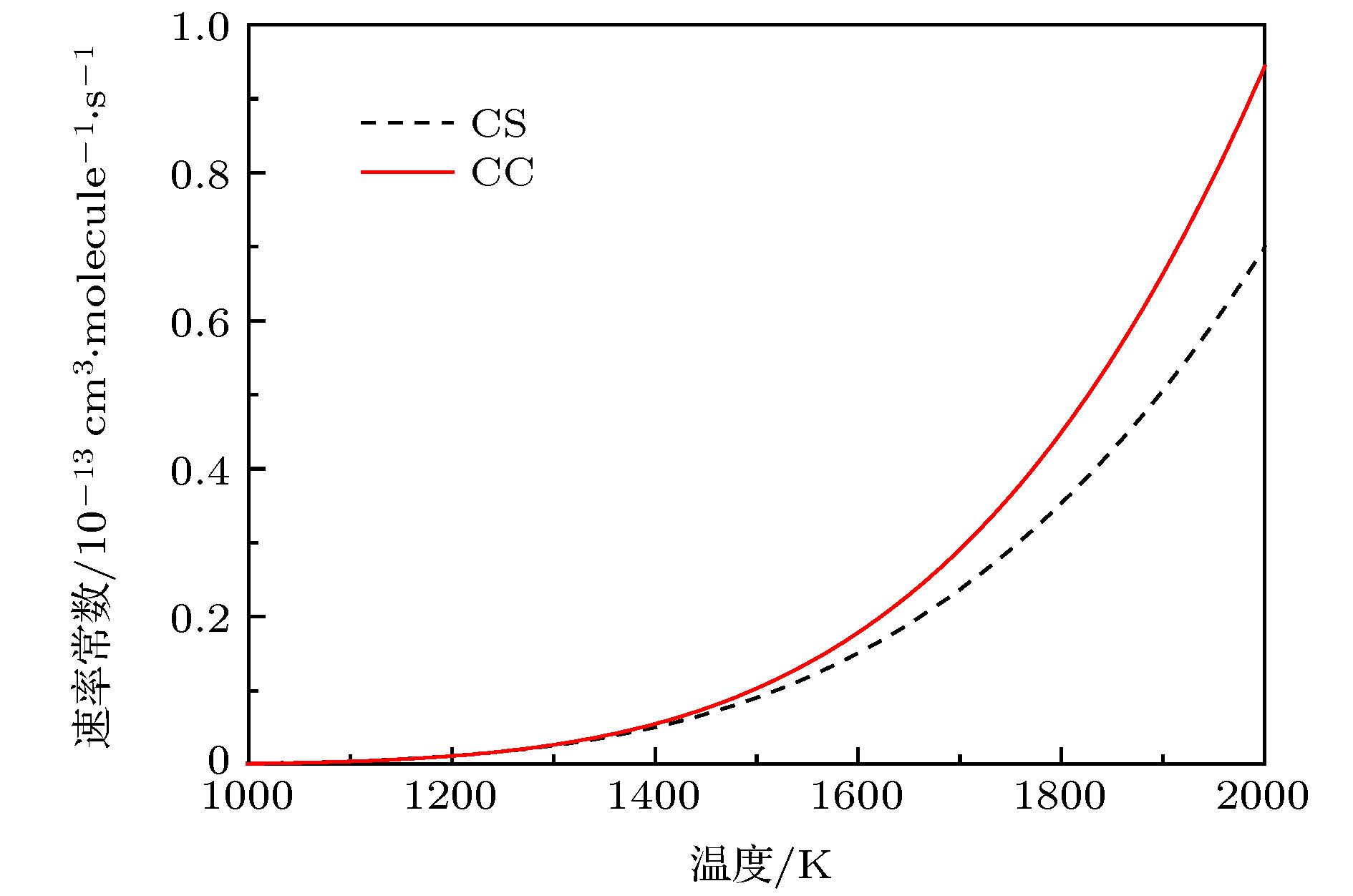
$(\tilde{\rm X}{}^3 {\rm A''})$ 势能面, 运用切比雪夫波包方法对初始态为($\nu = 0{\rm{ }},j = 0$ )的$ {\rm{C}}\left( {^3{\rm{P}}} \right) + $ $ {\rm H}_2 ({\rm X}^1\Sigma^+_{\rm g}) \to {\rm H} ({}^2{\rm S}) + {\rm CH}({}^2\Pi)$ 反应体系在1.0—2.0 eV 的碰撞能量范围内进行了动力学研究. 通过对角动量量子数J = 60以下的所有分波进行计算, 得到了反应几率、积分散射截面和速率常数. 计算中用到了耦合态近似方法和考虑科里奥利耦合效应的精确量子方法. 通过对比发现, 随着角动量量子数以及能量的增加, 科里奥利耦合效应的影响越发显著, 因而对于该反应体系, 科里奥利耦合效应不可忽略. 本文计算所得的积分散射截面和速率常数尚无实验数据可以比较, 对该反应的后续研究有一定的参考价值.The C(3P) + H2 → CH+H reaction in a collision energy range of 1.0–2.0 eV with the initial state$\nu = 0{\rm{ }},j = 0$ is investigated based on the new potential energy surface (PES) by using the Chebyshev wave packet method. All partial wave contributions up to J = 60 are calculated explicitly by the coupled state (CS) approximation method and the Coriolis coupling (CC) effect. Dynamic properties such as reaction probabilities, integral cross sections, and state specific rate constants are calculated. The calculated probabilities and integral reaction cross sections display an increasing trend with the increase of the collision energy and an oscillatory structure due to the CH2 well on the reaction path. The thermal rate constants of the endoergic reaction with a temperature ranging from 1000 K to 2000 K are obtained also. The calculated rate constants increase in the entire temperature range, showing a sharp T dependence in a range of 1400–2000 K. The rate constants are sensitive to the temperature due to the high threshold of the title reaction. In addition, the results of the exact calculations including CC effect are compared with those from the CS approximation. For smaller J, the CS probabilities are larger than the CC results, while for larger J, they are smaller than the CC ones. For reaction cross sections and rate constants, the CS results and the CC ones are in good agreement with each other at lower energy. However, they turn different at higher energy. The comparison between the CC and CS results indicates that neglecting the Coriolis coupling leads the cross sections and the rate constants to be underestimated due to the formation of a CH2 complex supported by stationary point of CH2(${\tilde{\rm X}}{}^3 \rm A''$ ) PES. It is suggested that the CH2 complex plays an important role in the process of the title reaction. However, it seems to overestimate the CS and CC rate constants because the barrier recrossing is neglected. Unfortunately, the results obtained in the present work have no corresponding theoretical or experimental data to be compared with, therefore these results provide simply a certain reference significance to the follow-up study of the title reaction.-
Keywords:
- CH2 system /
- potential energy surface /
- reaction probability /
- integral cross section
[1] Bockhorn H, Galdo N, Herbertz H A, Fetting F 1971 Combust. Sci. Technol. 2 329
 Google Scholar
Google Scholar
[2] Flower D R, Pineau des Foreêts G 1998 Mon. Not. R. Astron. Soc. 297 1182
Google Scholar
[3] Bucher M E, Glinski R J 1999 Mon. Not. R. Astron. Soc. 308 29
Google Scholar
[4] Bearda R A, vanHemert M C, vanDishoeck E F 1992 J. Chem. Phys. 97 8240
Google Scholar
[5] Scott D C, De Juan, J, Robie D C, Schwartz-Lavi D, Reisler H 1992 J. Phys. Chem. 96 2509
Google Scholar
[6] Jursich G M, Wiesenfeld J R 1985 J. Chem. Phys. 83 910
Google Scholar
[7] Mikulecky K, Gericke K H 1993 J. Chem. Phys. 98 1244
Google Scholar
[8] Bussery-Honvault B, Honvault P, Launay J M 2001 J. Chem. Phys. 115 10701
Google Scholar
[9] Bussery-Honvault B, Julien J, Honvault P, Launay J M 2005 Phys. Chem. Chem. Phys. 7 1476
Google Scholar
[10] Joseph S, Varandas A J C 2009 J. Phys. Chem. A 113 4175
Google Scholar
[11] Zhang C F, Fu M K, Shen Z T, Ma H T, Bian W S 2014 J. Chem. Phys. 140 234301
Google Scholar
[12] Lin S Y, Guo H 2004 J. Phys. Chem. A 108 2141
Google Scholar
[13] Sun Z P, Zhang C F, Lin S Y, Zheng Y J, Meng Q T, Bian W S 2013 J. Chem. Phys. 139 014306
Google Scholar
[14] Lu R F, Wang Y H, Deng K M 2013 J. Comput. Chem. 34 1735
Google Scholar
[15] Joseph S, Caridade P J S B, Varandas A J C 2011 J. Phys. Chem. A 115 7882
Google Scholar
[16] Wu Y, Zhang C F, Cao J W, Bian W S 2014 J. Phys. Chem. A 118 4235
Google Scholar
[17] Shen Z T, Cao J W, Bian W S 2015 J. Chem. Phys. 142 164309
Google Scholar
[18] Hickson K M, Suleimanov Y V 2017 Phys. Chem. Chem. Phys. 19 480
Google Scholar
[19] González-Lezana T, Larrégaray P, Bonnet L, Wu Y N, Bian W S 2018 J. Chem. Phys. 148 234305
Google Scholar
[20] Knowles P, Handy N C, Carter S 1983 Mol. Phys. 49 681
Google Scholar
[21] Murrell J, Dunne L 1983 Chem. Phys. Lett. 102 155
Google Scholar
[22] Harding L B, Guadagnini R, Schatz G C 1993 J. Phys. Chem. 97 5472
Google Scholar
[23] Werner H J, Knowles P J 1988 J. Chem. Phys. 89 5803
Google Scholar
[24] Knowles P J, Werner H J 1988 Chem. Phys. Lett. 145 514
Google Scholar
[25] van Harrevelt R, Van Hemert M C, Schatz G C 2002 J. Chem. Phys. 116 6002
Google Scholar
[26] Gamallo P, Defazio P, Akpinar S, Petrongolo C 2012 J. Phys. Chem. A 116 8291
Google Scholar
[27] Guadagnini R, Schatz G C 1996 J. Phys. Chem. 100 18944
Google Scholar
[28] Scholefield M R, Choi J H, Goyal S, Reisler H 1998 Chem. Phys. Lett. 288 487
Google Scholar
[29] Ehbrecht A, Kowalski A, Ottinger Ch 1998 Chem. Phys. Lett. 284 205
Google Scholar
[30] Zhang L L, Liu D, Yue D G, Song Y Z, Meng Q T 2020 J. Phys. B: At. Mol. Opt. Phys. 53 095202
Google Scholar
[31] Zhang J Z H 1999 Theory and Application of Quantum Molecular Dynamics (Singapore: World Scientific) pp201–218
[32] Gao F, Wang X L, Zhao W L, Song Y Z, Meng Q T 2018 Eur. Phys. J. D 72 224
Google Scholar
[33] Gao F, Zhang L L, Zhao W L, Meng Q T, Song Y Z 2019 J. Chem. Phys. 150 224304
Google Scholar
[34] Mandelshtam V A, Taylor H S 1995 J. Chem. Phys. 102 7390
Google Scholar
[35] Mandelshtam V A, Taylor H S 1995 J. Chem. Phys. 103 2903
Google Scholar
[36] Tal-Ezer H, Kosloff R 1984 J. Chem. Phys. 81 3967
Google Scholar
[37] Zhang D H, Zhang J Z H 1994 J. Chem. Phys. 101 1146
Google Scholar
[38] Meijer A J H M, Goldfield E M, Gray S K, Balint-Kurti G G 1998 Chem. Phys. Lett. 293 270
Google Scholar
[39] Althorpe S C 2001 J. Chem. Phys. 114 1601
Google Scholar
[40] Lin S Y, Guo H 2006 J. Chem. Phys. 124 031101
Google Scholar
[41] Blint R, Newton M 1975 Chem. Phys. Lett. 32 178
Google Scholar
[42] Zhai H C, Lin S Y 2015 Chem. Phys. 455 57
Google Scholar
[43] Carroll T E, Goldfield E M 2001 J. Phys. Chem. A 105 2251
Google Scholar
[44] Meijer A J H M, Goldeld E M 1999 J. Chem. Phys. 110 870
Google Scholar
[45] Chu T S, Han K L 2008 Phys. Chem. Chem. Phys. 10 2431
Google Scholar
[46] Peng Y, Jiang Z A, Chen J S 2017 J. Phys. Chem. A 121 2209
Google Scholar
-
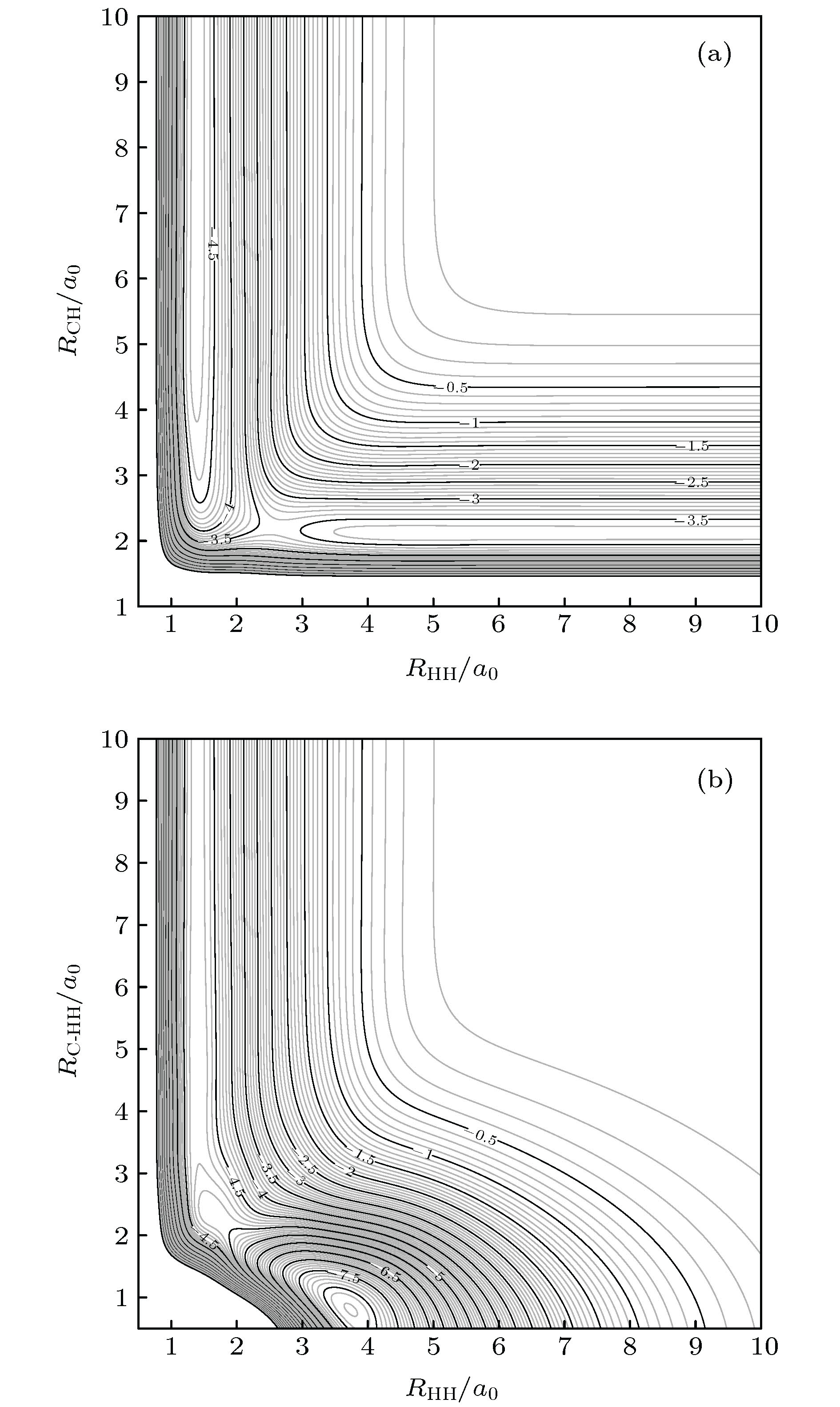
图 1 CH2等势线, 图中等势线间隔为0.1 eV (a) C沿着共线构型靠近H2分子; (b) C 沿着T构型插入H2
Fig. 1. Equipotential contour plot for CH2, the contour increments are 0.1 eV: (a) For bond stretching in C-H-H linear geometry; (b) for T-shaped insertion of C into H2 diatoms.
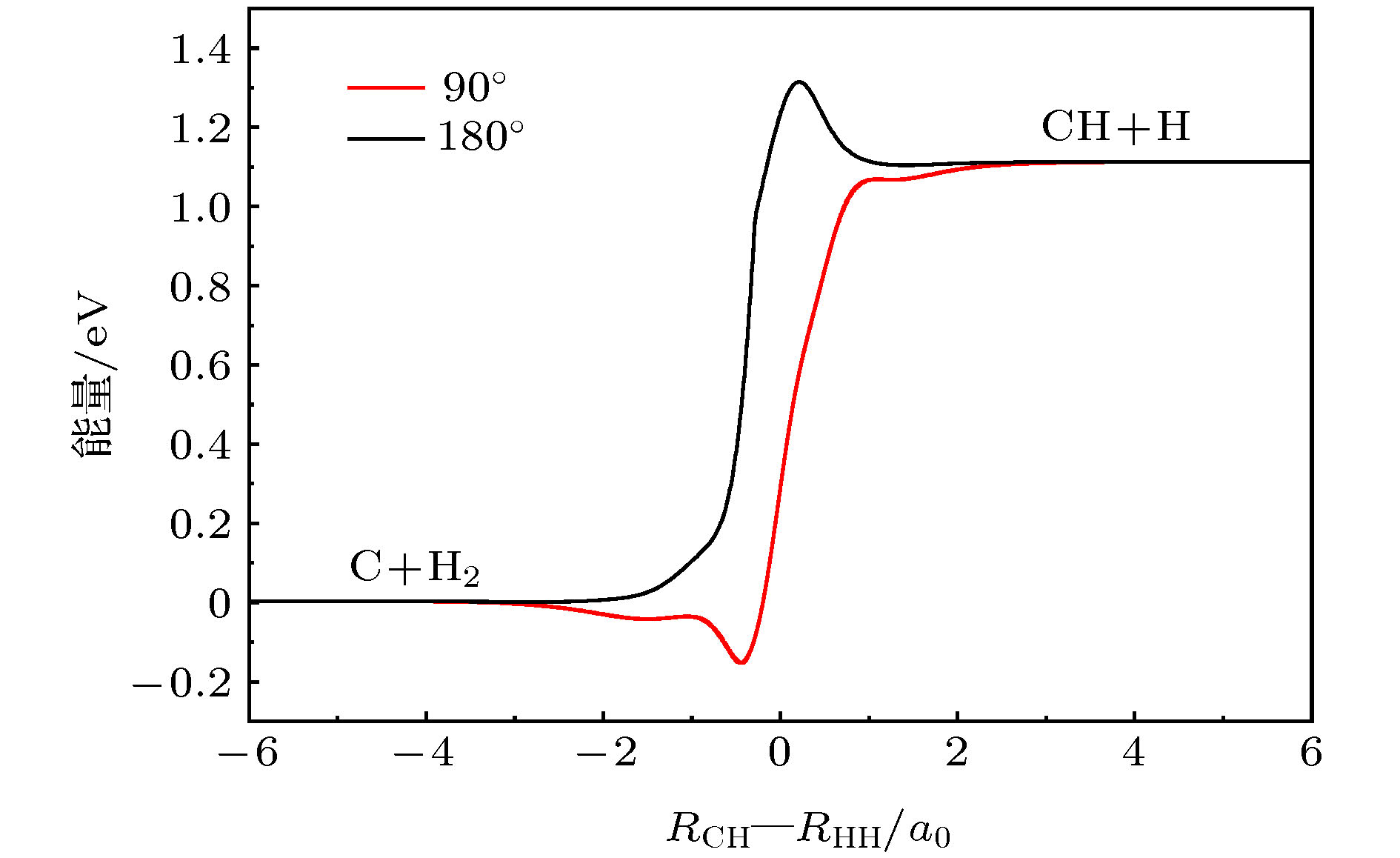
图 2 90°和180°的最小能量路径
Fig. 2. The minimum energy paths as a function of RCH-RHH at 90° and 180°.
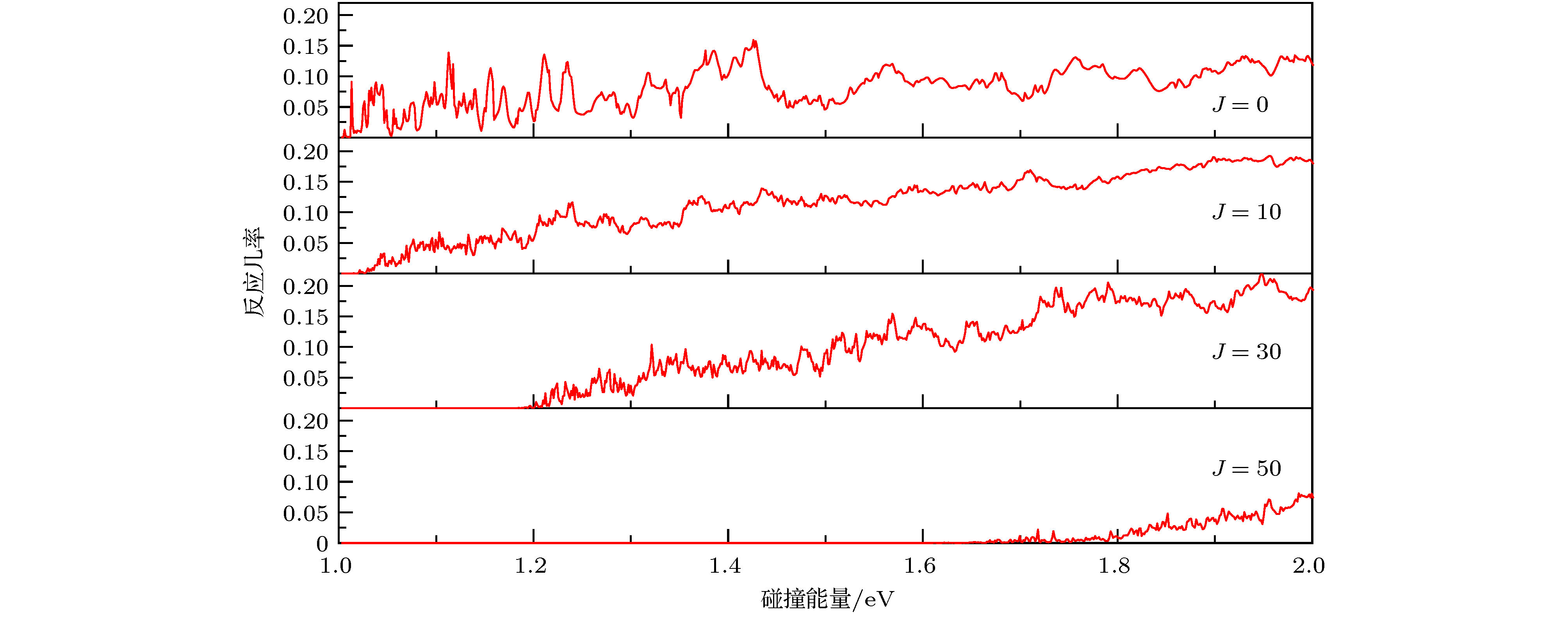
图 3 不同分波的反应几率随着碰撞能量的变化
Fig. 3. The reaction probabilities vs. collision energy at different J.
表 1 波包计算中的数值参量(除特殊说明, 均采用原子单位a.u.)
Table 1. Parameters used in wave packet calculation (The atomic unit is used in the calculation unless otherwise stated).
坐标取值范围
和基组数$R \in ({10^{{\rm{ - }}16}}, \, 16)$, $({N_R} = 203)$ $r \in (0.5, \, 12)$, $({N_r} = 99)$ $\gamma \in ({90^ \circ }, \, {180^ \circ })$, $({N_\gamma } = 50)$ 吸收势 ${R_{\rm{d}}} = 11.0$, ${d_R} = 0.0006$ ${r_{\rm{d}}} = 7.5$, ${d_r} = 0.001$ 初始波包 ${R_0} = 8.0$, ${E_0} = 1.55\;{\rm{ eV}}$, $\delta = 0.3$ 光谱控制 0.5 流计算的位置 ${r_{\rm{f}}} = 7.4$ 传播步数 100000  下载: 导出CSV
下载: 导出CSV
-
[1] Bockhorn H, Galdo N, Herbertz H A, Fetting F 1971 Combust. Sci. Technol. 2 329
Google Scholar
[2] Flower D R, Pineau des Foreêts G 1998 Mon. Not. R. Astron. Soc. 297 1182
Google Scholar
[3] Bucher M E, Glinski R J 1999 Mon. Not. R. Astron. Soc. 308 29
Google Scholar
[4] Bearda R A, vanHemert M C, vanDishoeck E F 1992 J. Chem. Phys. 97 8240
Google Scholar
[5] Scott D C, De Juan, J, Robie D C, Schwartz-Lavi D, Reisler H 1992 J. Phys. Chem. 96 2509
Google Scholar
[6] Jursich G M, Wiesenfeld J R 1985 J. Chem. Phys. 83 910
Google Scholar
[7] Mikulecky K, Gericke K H 1993 J. Chem. Phys. 98 1244
Google Scholar
[8] Bussery-Honvault B, Honvault P, Launay J M 2001 J. Chem. Phys. 115 10701
Google Scholar
[9] Bussery-Honvault B, Julien J, Honvault P, Launay J M 2005 Phys. Chem. Chem. Phys. 7 1476
Google Scholar
[10] Joseph S, Varandas A J C 2009 J. Phys. Chem. A 113 4175
Google Scholar
[11] Zhang C F, Fu M K, Shen Z T, Ma H T, Bian W S 2014 J. Chem. Phys. 140 234301
Google Scholar
[12] Lin S Y, Guo H 2004 J. Phys. Chem. A 108 2141
Google Scholar
[13] Sun Z P, Zhang C F, Lin S Y, Zheng Y J, Meng Q T, Bian W S 2013 J. Chem. Phys. 139 014306
Google Scholar
[14] Lu R F, Wang Y H, Deng K M 2013 J. Comput. Chem. 34 1735
Google Scholar
[15] Joseph S, Caridade P J S B, Varandas A J C 2011 J. Phys. Chem. A 115 7882
Google Scholar
[16] Wu Y, Zhang C F, Cao J W, Bian W S 2014 J. Phys. Chem. A 118 4235
Google Scholar
[17] Shen Z T, Cao J W, Bian W S 2015 J. Chem. Phys. 142 164309
Google Scholar
[18] Hickson K M, Suleimanov Y V 2017 Phys. Chem. Chem. Phys. 19 480
Google Scholar
[19] González-Lezana T, Larrégaray P, Bonnet L, Wu Y N, Bian W S 2018 J. Chem. Phys. 148 234305
Google Scholar
[20] Knowles P, Handy N C, Carter S 1983 Mol. Phys. 49 681
Google Scholar
[21] Murrell J, Dunne L 1983 Chem. Phys. Lett. 102 155
Google Scholar
[22] Harding L B, Guadagnini R, Schatz G C 1993 J. Phys. Chem. 97 5472
Google Scholar
[23] Werner H J, Knowles P J 1988 J. Chem. Phys. 89 5803
Google Scholar
[24] Knowles P J, Werner H J 1988 Chem. Phys. Lett. 145 514
Google Scholar
[25] van Harrevelt R, Van Hemert M C, Schatz G C 2002 J. Chem. Phys. 116 6002
Google Scholar
[26] Gamallo P, Defazio P, Akpinar S, Petrongolo C 2012 J. Phys. Chem. A 116 8291
Google Scholar
[27] Guadagnini R, Schatz G C 1996 J. Phys. Chem. 100 18944
Google Scholar
[28] Scholefield M R, Choi J H, Goyal S, Reisler H 1998 Chem. Phys. Lett. 288 487
Google Scholar
[29] Ehbrecht A, Kowalski A, Ottinger Ch 1998 Chem. Phys. Lett. 284 205
Google Scholar
[30] Zhang L L, Liu D, Yue D G, Song Y Z, Meng Q T 2020 J. Phys. B: At. Mol. Opt. Phys. 53 095202
Google Scholar
[31] Zhang J Z H 1999 Theory and Application of Quantum Molecular Dynamics (Singapore: World Scientific) pp201–218
[32] Gao F, Wang X L, Zhao W L, Song Y Z, Meng Q T 2018 Eur. Phys. J. D 72 224
Google Scholar
[33] Gao F, Zhang L L, Zhao W L, Meng Q T, Song Y Z 2019 J. Chem. Phys. 150 224304
Google Scholar
[34] Mandelshtam V A, Taylor H S 1995 J. Chem. Phys. 102 7390
Google Scholar
[35] Mandelshtam V A, Taylor H S 1995 J. Chem. Phys. 103 2903
Google Scholar
[36] Tal-Ezer H, Kosloff R 1984 J. Chem. Phys. 81 3967
Google Scholar
[37] Zhang D H, Zhang J Z H 1994 J. Chem. Phys. 101 1146
Google Scholar
[38] Meijer A J H M, Goldfield E M, Gray S K, Balint-Kurti G G 1998 Chem. Phys. Lett. 293 270
Google Scholar
[39] Althorpe S C 2001 J. Chem. Phys. 114 1601
Google Scholar
[40] Lin S Y, Guo H 2006 J. Chem. Phys. 124 031101
Google Scholar
[41] Blint R, Newton M 1975 Chem. Phys. Lett. 32 178
Google Scholar
[42] Zhai H C, Lin S Y 2015 Chem. Phys. 455 57
Google Scholar
[43] Carroll T E, Goldfield E M 2001 J. Phys. Chem. A 105 2251
Google Scholar
[44] Meijer A J H M, Goldeld E M 1999 J. Chem. Phys. 110 870
Google Scholar
[45] Chu T S, Han K L 2008 Phys. Chem. Chem. Phys. 10 2431
Google Scholar
[46] Peng Y, Jiang Z A, Chen J S 2017 J. Phys. Chem. A 121 2209
Google Scholar
-
[1] 赵文丽, 宋玉志, 马超, 高峰, 孟庆田. 基于一个新SiH2(11A′)势能面的H+SiH反应动力学研究. 物理学报, 2024, 73(20): 203401. doi: 10.7498/aps.73.20240859 [2] 崔子纯, 杨莫涵, 阮晓鹏, 范晓丽, 周峰, 刘维民. 高通量计算二维材料界面摩擦. 物理学报, 2023, 72(2): 026801. doi: 10.7498/aps.72.20221676 [3] 邱梓恒, AhmedYousif Ghazal, 龙金友, 张嵩. 三乙胺分子构象与红外光谱的理论研究. 物理学报, 2022, 71(10): 103601. doi: 10.7498/aps.71.20220123 [4] 赵文丽, 孙丰伟, 张红, 王永刚, 高峰, 孟庆田. $ {\text{D}} + {\text{Si}}{{\text{D}}^ + } \to {{\text{D}}_2} + {\text{S}}{{\text{i}}^ + } $ 反应量子波包动力学研究. 物理学报, 2022, 71(22): 228201. doi: 10.7498/aps.71.20221155[5] 李文涛, 袁美玲, 王杰敏. C++H2反应的动力学研究: 基于一个新构建的势能面. 物理学报, 2022, 71(9): 093402. doi: 10.7498/aps.71.20212241 [6] 乔政, 王雅丽, 吴明伟, 凤尔银, 黄武英. Xe-NH (X3∑-)体系的势能面和冷碰撞动力学研究. 物理学报, 2018, 67(21): 213401. doi: 10.7498/aps.67.20181321 [7] 魏强. 基于4A势能面研究C(3P)+NO(X2)CO(X1+)+N(4S)反应的立体动力学性质. 物理学报, 2015, 64(17): 173401. doi: 10.7498/aps.64.173401 [8] 杨雪, 闫冰, 连科研, 丁大军. 1,2-环己二酮基态光解离反应的理论研究. 物理学报, 2015, 64(21): 213101. doi: 10.7498/aps.64.213101 [9] 韩玉龙, 李真, 汪江洪, 凤尔银, 黄武英. Mg-CO体系的相互作用势和光谱预测. 物理学报, 2013, 62(9): 093101. doi: 10.7498/aps.62.093101 [10] 沈光先, 汪荣凯, 令狐荣锋, 周勋, 杨向东. He-HD (HT, DT) 非对称碰撞体系振转势能面及微分散射截面的理论计算. 物理学报, 2012, 61(21): 213101. doi: 10.7498/aps.61.213101 [11] 臧华平, 李文峰, 令狐荣锋, 程新路, 杨向东. 钠分子同位素替代对低温下的He-Na2冷碰撞体系转动激发积分散射截面的影响. 物理学报, 2011, 60(2): 020304. doi: 10.7498/aps.60.020304 [12] 李文峰, 令狐荣锋, 程新路, 杨向东. 氦同位素原子与钠分子碰撞转动激发积分散射截面的理论计算. 物理学报, 2010, 59(7): 4591-4597. doi: 10.7498/aps.59.4591 [13] 余春日, 汪荣凯, 程新路, 杨向东. He-HF体系势能模型对散射截面影响的理论研究. 物理学报, 2007, 56(5): 2577-2584. doi: 10.7498/aps.56.2577 [14] 余春日, 凤尔银, 程新路, 杨向东. He-HI复合物势能面及微分散射截面的理论研究. 物理学报, 2007, 56(8): 4441-4447. doi: 10.7498/aps.56.4441 [15] 姜振益, 李盛涛. NiTi合金势能面的第一性原理研究. 物理学报, 2006, 55(11): 6032-6035. doi: 10.7498/aps.55.6032 [16] 韩慧仙, 彭 谦, 文振翼, 王育彬. S2O分子的局域势能面和振动光谱的解析. 物理学报, 2005, 54(1): 78-84. doi: 10.7498/aps.54.78 [17] 王晓艳, 丁世良. 用李代数方法构造四原子分子的势能面. 物理学报, 2004, 53(2): 423-426. doi: 10.7498/aps.53.423 [18] 张程华, 邱 巍, 辛俊丽, 牛英煜, 王晓伟, 王京阳. 电子碰撞下氢原子单离化反应三重微分散射截面的计算. 物理学报, 2003, 52(10): 2449-2452. doi: 10.7498/aps.52.2449 [19] 孙桂华, 杨向东. H+H2反应截面的全量子力学研究. 物理学报, 2002, 51(3): 506-511. doi: 10.7498/aps.51.506 [20] 金石琦, 徐至展. e-Ar(3p54s,J=2)的微分散射截面. 物理学报, 1998, 47(10): 1621-1624. doi: 10.7498/aps.47.1621







 下载:
下载:

计量
- 文章访问数: 6572
- PDF下载量: 74
- 被引次数: 0
