










-
三价稀土铒离子(Er3+)掺杂二氧化钛(TiO2)因其优异的光电性能, 在众多稀土掺杂发光晶体材料中脱颖而出, 具有非常广泛的应用前景. 利用CALYPSO (Crystal structure AnaLYsis by Particle Swarm Optimization)结构搜索方法和第一性原理计算, 成功地预测和探究了三价铒离子掺杂二氧化钛(Er3+:TiO2)晶体的基态结构. 首次报道Er3+掺杂TiO2的最低能量结构具有
$P\overline 4 m2$ 空间对称性. 当Er3+离子掺杂基质晶体时, Er3+离子占据Ti4+离子的位置, 并造成结构畸变, 最终使得Er3+离子的局域对称性由D2d降低为C2v. 通过电子结构计算发现Er3+掺杂TiO2的能带隙值约为2.27 eV, 这表明Er3+离子的掺杂会导致体系的带隙值降低, 但没有改变其半导体性质, 从而在光伏电池和半导体激光器等领域具有更广泛的应用. 这些发现不仅为进一步探索Er3+:TiO2晶体的性质和应用提供了数据参考, 也为探究其他稀土掺杂晶体材料提供了最新的方法.-
关键词:
- CALYPSO /
- Er3+掺杂TiO2 /
- 局域结构 /
- 第一性原理计算
Trivalent rare earth erbium ion (Er3+) doped titanium oxide (TiO2) can possess a very wide range of applications due to its excellent optoelectronic properties, thus standing out among many rare-earth-doped luminescent crystals. However, the issues regarding local structure and electronic properties have not been finalized. To address these problems, the CALYPSO (Crystal structure AnaLYsis by Particle Swarm Optimization) method combined with the first-principles calculations is employed, and many converged structures of Er3+-doped TiO2 are successfully obtained. Further structural optimization is performed by using the VASP (Vienna ab initio simulation package) software package, and we report for the first time that the lowest energy structure of Er3+-doped TiO2 has the$ P\overline 4 $ m2 symmetry. It can be observed that the doped Er3+ ions enter into the host crystal and occupy the positions of Ti4+ ions, resulting in structural distortion, which eventually leads the local Er3+ coordination site symmetry to reduce from D2d into C2v. We speculate that there are two reasons: 1) the difference in charge between Er3+ ions and Ti4+ ions leads to charge compensation; 2) the difference between their electron radii is obvious: the radius is 0.0881 for Er3+ ion and 0.0881 for Ti4+ ion. In addition, during the structural search, we also find many metastable structures that may exist at a special temperature or pressure, which play an important role in the studying of structural evolution. When the electronic band structure of the Er3+-doped TiO2 system is calculated, we adopt the method of local density approximation (LDA) combined with the on-site Coulomb repulsion parameter U to accurately describe the strongly correlated system. For the specific value of U, we adopt 3.5 eV and 7.6 eV to describe the strong correlation of 3d electrons of Ti4+ ions and 4f electrons of Er3+ ions, respectively. According to the calculation of electronic properties, the band gap value of Er3+ doped TiO2 is about 2.27 eV, which is lower than that of the host crystal (Eg = 2.40 eV). The results show that the reduction in the band gap is mainly caused by the f state of Er3+ ions. The doping of Er ion does reduce the band gap value, but it does not change the conductivity of the system, which have great application prospect in diode-pumped laser. These findings not only provide the data for further exploring the properties and applications of Er3+:TiO2 crystals, but also present an approach to studying other rare-earth-doped crystalline materials.-
Keywords:
- CALYPSO /
- Er3+-doped TiO2 /
- local structure /
- first-principles calculations
[1] 钟淑琳, 仇家豪, 罗文崴, 吴木生 2021 物理学报 70 158203
 Google Scholar
Google Scholar
Zhong S L, Qiu J H, Luo W W, Wu M S 2021 Acta Phys. Sin. 70 158203
Google Scholar
[2] 孟勇军, 李洪, 唐建伟, 陈学文 2022 物理学报 71 027801
Google Scholar
Meng Y J, Li H, Tang J W, Chen X W 2022 Acta Phys. Sin. 71 027801
Google Scholar
[3] Wu B, Zhao L, Wang Y, Dong H, Yu H 2019 RSC Adv. 9 42228
Google Scholar
[4] Menezes L D S, Araújo C B D 2015 J. Braz. Chem. Soc. 26 2405
[5] Stengl V, Bakardjieva S, Murafa N 2009 Mater. Chem. Phys. 114 217
Google Scholar
[6] Hassan M S, Amna T, Yang O B, Kim H C, Khil M S 2012 Ceram. Int. 38 5925
Google Scholar
[7] Borlaf M, Colomer M T, Moreno R, Ortiz A L 2014 J. Eur. Ceram. Soc. 34 4457
Google Scholar
[8] Bao R, Li R, Chen C, Wu H, Xia J, Long C, Li H 2019 J. Phys. Chem. Solid. 126 78
Google Scholar
[9] Li J G, Wang X H, Kamiyama H, Ishigaki T, Sekiguchi T 2006 Thin Solid Films 506 292
[10] Agrios A G, Pochat P 2005 J. Appl. Electrochem. 35 655
Google Scholar
[11] Camps I, Borlaf M, Toudert J, Andres A D, Colomer M T, Moreno R, Serna R 2018 J. Alloys Compd. 735 2267
Google Scholar
[12] Talane T E 2018 M. S. Thesis (Gauteng Province: University of South Africa)
[13] Pablo L I, Laeticia P, Jonathan M, Davide J, Nadia G B, Diego P, Sonia F, Chiara N, Fabrizio G, Daniel M 2017 J. Non-Cryst. Solids 460 161
Google Scholar
[14] Mignotte C 2004 Appl. Surf. Sci. 226 355
Google Scholar
[15] Talane T E, Mbule P S, Noto L L, Shingange K, Mhlongo G H, Mothudi B M, Dhlamini M S 2018 Mater. Res. Bull. 108 234
Google Scholar
[16] Wild J D, Meijerink A, Rath J K, van Sark W G J H M, Schropp R E I 2011 Energy Environ. Sci. 4 4835
Google Scholar
[17] Pablo L I, Diego P, Nadia G B, Davide J, Giovanni B, Laeticia P, Daniel M 2018 Nanomaterials 8 20
Google Scholar
[18] van den Hoven G N, Koper R J I M, Polman A, Dam C V, Uffelen J W M V, Smit M K 1996 Appl. Phys. Lett. 68 1886
Google Scholar
[19] Jia C W, Zhao J G, Duan H G, Xie E Q 2007 Mater. Lett. 61 4389
Google Scholar
[20] Fu C Y, Liao J S, Luo W Q, Li R F, Chen X Y 2008 Opt. Lett. 33 953
Google Scholar
[21] Luo W Q, Fu C Y, Li R F, Liu Y S, Zhu H M, Chen X Y 2011 Small 7 3046
Google Scholar
[22] Ren Z, Wu J, Wang N, Li X 2018 J. Mater. Chem. A 6 15348
Google Scholar
[23] Mazierski P, Mikolajczyk A, Grzybd T, Caicedo P N A, Wei Z, Kowalska E, Henry P P, Adriana Z M, Nadolna J 2020 Appl. Surf. Sci. 527 146815
Google Scholar
[24] Wang Y C, Lv J, Zhu L, Ma Y M 2012 Comput. Phys. Commun. 183 2063
Google Scholar
[25] Wang Y C, Lv J, Zhu L, Lu S H, Yin K T, Li Q, Wang H, Zhang L J, Ma Y M 2015 J. Phys. Condens. Matter. 27 203203
Google Scholar
[26] Wang H, Wang Y C, Lv J, Li Q, Zhang L J, Ma Y M 2016 Comput. Mater. Sci. 112 406
Google Scholar
[27] Wang Y C, Lv J, Zhu L, Ma Y M 2010 Phys. Rev. B 82 094116
Google Scholar
[28] Gao B, Gao P, Lu S, Lv J, Wang Y, Ma Y 2019 Sci. Bull. 64 301
Google Scholar
[29] Wang Y, Miao M, Lv J, Zhu L, Yin K, Liu H, Ma Y 2012 J. Chem. Phys. 137 224108
Google Scholar
[30] Hafner J 2008 J. Comput. Chem. 29 2044
Google Scholar
[31] Kresse G, Furthmüller J 1996 Phys. Rev. B 54 11169
Google Scholar
[32] Perdew J P, Burke K, Ernzerhof M 1996 Phys. Rev. Lett. 77 3865
Google Scholar
[33] Sanna S, Schmidt W G, Frauenheim T, Gerstmann U 2009 Phys. Rev. B 80 104120
Google Scholar
[34] Xiao Y, Ju M, Yuan H K, Yeung Y Y 2021 J. Phys. Chem. C 125 18015
Google Scholar
[35] Togo A, Tanaka I 2015 Scr. Mater. 108 1
Google Scholar
[36] Phenicie C M, Stevenson P, Welinski S, Rose B C, Asfaw A T, Cava R J, Lyon S A, de Leon N P, Thompson J D 2019 Nano Lett. 19 8928
Google Scholar
[37] Mills A, Hunte S L 1997 J. Photochem. Photobiol. A 108 1
Google Scholar
[38] Yang J, Hu Y, Jin C, Zhuge L, Wu X 2017 Thin Solid Films 637 9
Google Scholar
[39] Pan L, Xiao Y, Kuang X Y, Ju M 2021 Mater. Chem. Phys. 257 123824
Google Scholar
[40] Savin A, Nesper R, Wengert S, Fässler T F 1997 Angew. Chem. Int. Ed. Engl. 36 1808
Google Scholar
[41] Lu T, Chen F 2011 Acta Phys. Chim. Sin. 27 2786
Google Scholar
[42] Fuentealba P, Chamorro E, Santos J C 2007 Theoretical Aspects of Chemical Reactivity 19 57
-

图 1 通过CALYPSO结构搜索法确定纯TiO2 (a)和Er3+掺杂TiO2 (b)的晶体结构
Fig. 1. Crystal structures of the pure TiO2 (a) and Er3+-doped TiO2 (b) by the CALYPSO structure search method.

图 2 Er3+:TiO2晶体的亚稳态结构
Fig. 2. Coordination structures of the metastable for Er3+:TiO2.
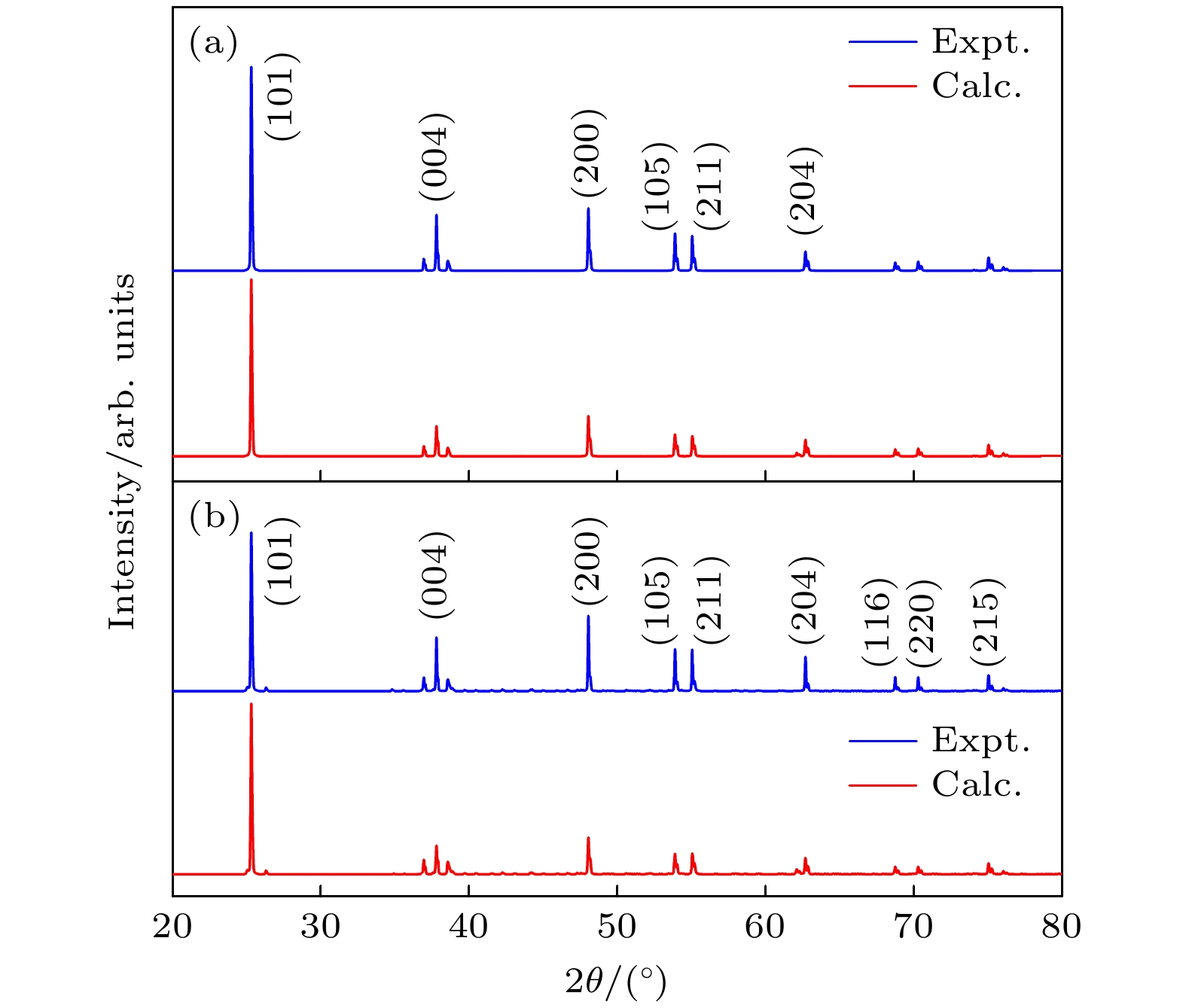
图 3 模拟的(a) TiO2和(b) Er3+掺杂TiO2的XRD图, 并与实验值进行对比
Fig. 3. Simulated X-ray diffraction patterns of (a) TiO2 and (b) Er3+-doped TiO2 compared with experimental data.

图 4 计算的(a) TiO2和(b) Er3+掺杂TiO2的声子谱
Fig. 4. Calculated phonon spectra of the (a) TiO2 and (b) Er3+-doped TiO2.
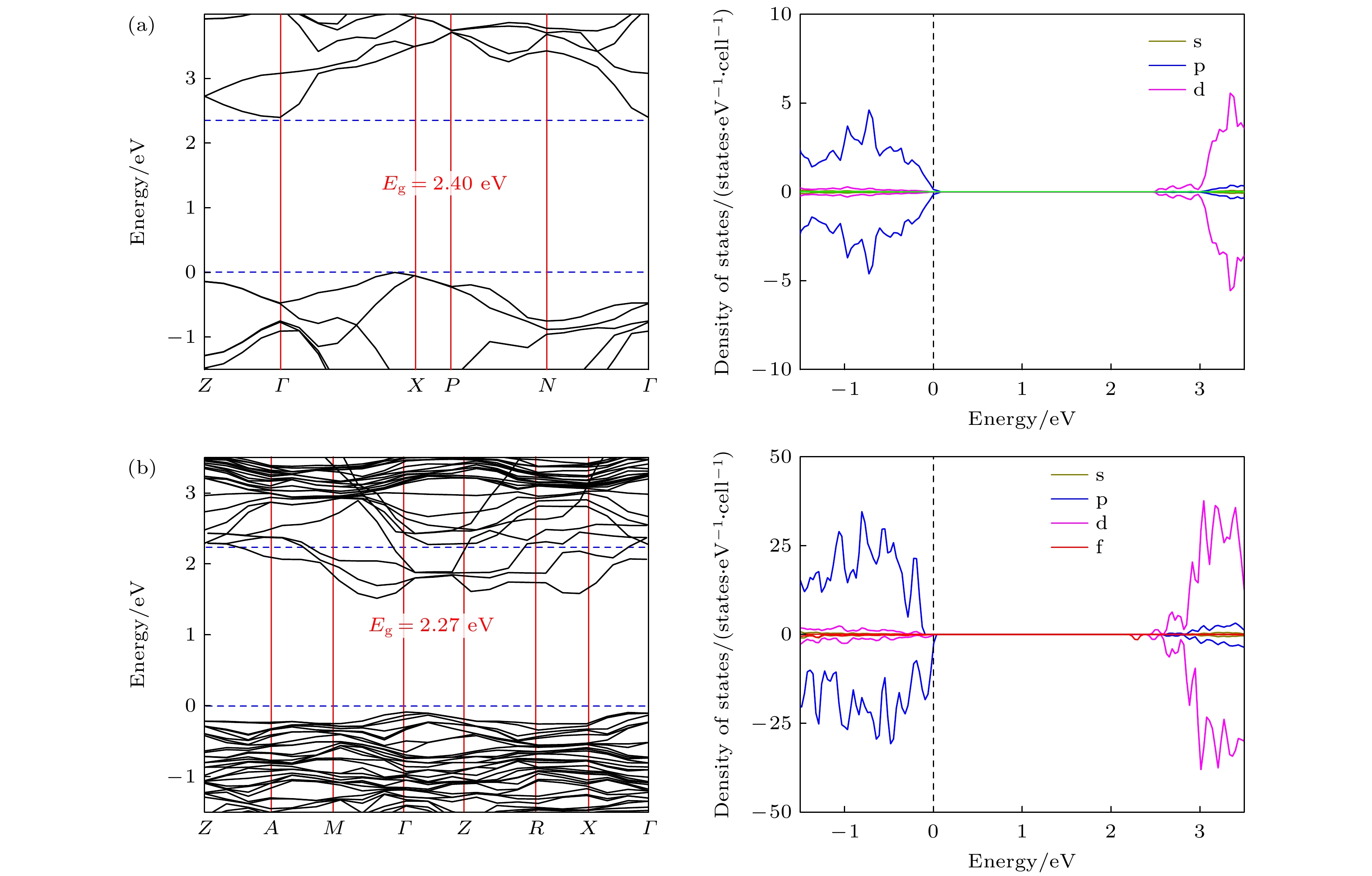
图 5 采用LDA方法计算的(a) TiO2和(b) Er3+掺杂TiO2的能带结构和态密度
Fig. 5. Band structures and the DOS of (a) TiO2 and (b) Er3+-doped TiO2, all calculated by the LDA method.

图 6 计算得到的Er3+:TiO2电子局域函数 (a)基态结构; (b) (010)平面
Fig. 6. Electron localized function of the Er3+:TiO2: (a) Ground-state structure; (b) (010) plane.
表 1 Er3+:TiO2能量最低结构中所有原子的坐标
Table 1. Coordinates of all atoms for the low-energy structure of Er3+:TiO2.
Atom x y z Wyckoff site
symmetryO(1) 0.247 0.249 0.204 4j O(2) 0.753 0.751 0.204 4k O(3) 0.249 0.753 0.796 2g O(4) 0.751 0.247 0.796 8l O(6) 0.753 0.249 0.204 2g O(7) 0.250 0.247 0.796 4k O(8) 0.751 0.753 0.796 4j O(11) 0.250 0.000 0.545 2f O(19) 0.500 0.753 0.036 2e Ti(1) 0.000 0.250 0.251 4j Ti(2) 0.000 0.750 0.251 4k Ti(3) 0.250 0.000 0.749 2g Ti(4) 0.750 0.000 0.749 4h Ti(10) 0.500 0.000 0.495 1d Er(1) 0.500 0.500 0.500 1c  下载: 导出CSV
下载: 导出CSV
表 2 Er3+:TiO2的基态结构以及亚稳态结构的晶格参数a, b, c, 原胞体积V, 相对能量∆E
Table 2. Structural parameters a, b and c, unit-cell volume, relative energies for the optimized TiO2 and metastable Er3+:TiO2
Space group a/Å b/Å c/Å V/Å3 ∆E/eV TiO2 I41/amd 7.568 7.568 9.515 545.003 — Er3+:TiO2 $ P\overline 4 $m2 7.682 7.682 9.798 578.193 0 Isomer (a) $ P\overline 4 $m2 7.681 7.681 9.799 578.213 0.051 Isomer (b) Cmmm 13.258 13.258 6.013 1051.764 0.853 Isomer (c) Cmmm 13.261 13.261 6.009 1051.988 0.975
下载: 导出CSV
-
[1] 钟淑琳, 仇家豪, 罗文崴, 吴木生 2021 物理学报 70 158203
Google Scholar
Zhong S L, Qiu J H, Luo W W, Wu M S 2021 Acta Phys. Sin. 70 158203
Google Scholar
[2] 孟勇军, 李洪, 唐建伟, 陈学文 2022 物理学报 71 027801
Google Scholar
Meng Y J, Li H, Tang J W, Chen X W 2022 Acta Phys. Sin. 71 027801
Google Scholar
[3] Wu B, Zhao L, Wang Y, Dong H, Yu H 2019 RSC Adv. 9 42228
Google Scholar
[4] Menezes L D S, Araújo C B D 2015 J. Braz. Chem. Soc. 26 2405
[5] Stengl V, Bakardjieva S, Murafa N 2009 Mater. Chem. Phys. 114 217
Google Scholar
[6] Hassan M S, Amna T, Yang O B, Kim H C, Khil M S 2012 Ceram. Int. 38 5925
Google Scholar
[7] Borlaf M, Colomer M T, Moreno R, Ortiz A L 2014 J. Eur. Ceram. Soc. 34 4457
Google Scholar
[8] Bao R, Li R, Chen C, Wu H, Xia J, Long C, Li H 2019 J. Phys. Chem. Solid. 126 78
Google Scholar
[9] Li J G, Wang X H, Kamiyama H, Ishigaki T, Sekiguchi T 2006 Thin Solid Films 506 292
[10] Agrios A G, Pochat P 2005 J. Appl. Electrochem. 35 655
Google Scholar
[11] Camps I, Borlaf M, Toudert J, Andres A D, Colomer M T, Moreno R, Serna R 2018 J. Alloys Compd. 735 2267
Google Scholar
[12] Talane T E 2018 M. S. Thesis (Gauteng Province: University of South Africa)
[13] Pablo L I, Laeticia P, Jonathan M, Davide J, Nadia G B, Diego P, Sonia F, Chiara N, Fabrizio G, Daniel M 2017 J. Non-Cryst. Solids 460 161
Google Scholar
[14] Mignotte C 2004 Appl. Surf. Sci. 226 355
Google Scholar
[15] Talane T E, Mbule P S, Noto L L, Shingange K, Mhlongo G H, Mothudi B M, Dhlamini M S 2018 Mater. Res. Bull. 108 234
Google Scholar
[16] Wild J D, Meijerink A, Rath J K, van Sark W G J H M, Schropp R E I 2011 Energy Environ. Sci. 4 4835
Google Scholar
[17] Pablo L I, Diego P, Nadia G B, Davide J, Giovanni B, Laeticia P, Daniel M 2018 Nanomaterials 8 20
Google Scholar
[18] van den Hoven G N, Koper R J I M, Polman A, Dam C V, Uffelen J W M V, Smit M K 1996 Appl. Phys. Lett. 68 1886
Google Scholar
[19] Jia C W, Zhao J G, Duan H G, Xie E Q 2007 Mater. Lett. 61 4389
Google Scholar
[20] Fu C Y, Liao J S, Luo W Q, Li R F, Chen X Y 2008 Opt. Lett. 33 953
Google Scholar
[21] Luo W Q, Fu C Y, Li R F, Liu Y S, Zhu H M, Chen X Y 2011 Small 7 3046
Google Scholar
[22] Ren Z, Wu J, Wang N, Li X 2018 J. Mater. Chem. A 6 15348
Google Scholar
[23] Mazierski P, Mikolajczyk A, Grzybd T, Caicedo P N A, Wei Z, Kowalska E, Henry P P, Adriana Z M, Nadolna J 2020 Appl. Surf. Sci. 527 146815
Google Scholar
[24] Wang Y C, Lv J, Zhu L, Ma Y M 2012 Comput. Phys. Commun. 183 2063
Google Scholar
[25] Wang Y C, Lv J, Zhu L, Lu S H, Yin K T, Li Q, Wang H, Zhang L J, Ma Y M 2015 J. Phys. Condens. Matter. 27 203203
Google Scholar
[26] Wang H, Wang Y C, Lv J, Li Q, Zhang L J, Ma Y M 2016 Comput. Mater. Sci. 112 406
Google Scholar
[27] Wang Y C, Lv J, Zhu L, Ma Y M 2010 Phys. Rev. B 82 094116
Google Scholar
[28] Gao B, Gao P, Lu S, Lv J, Wang Y, Ma Y 2019 Sci. Bull. 64 301
Google Scholar
[29] Wang Y, Miao M, Lv J, Zhu L, Yin K, Liu H, Ma Y 2012 J. Chem. Phys. 137 224108
Google Scholar
[30] Hafner J 2008 J. Comput. Chem. 29 2044
Google Scholar
[31] Kresse G, Furthmüller J 1996 Phys. Rev. B 54 11169
Google Scholar
[32] Perdew J P, Burke K, Ernzerhof M 1996 Phys. Rev. Lett. 77 3865
Google Scholar
[33] Sanna S, Schmidt W G, Frauenheim T, Gerstmann U 2009 Phys. Rev. B 80 104120
Google Scholar
[34] Xiao Y, Ju M, Yuan H K, Yeung Y Y 2021 J. Phys. Chem. C 125 18015
Google Scholar
[35] Togo A, Tanaka I 2015 Scr. Mater. 108 1
Google Scholar
[36] Phenicie C M, Stevenson P, Welinski S, Rose B C, Asfaw A T, Cava R J, Lyon S A, de Leon N P, Thompson J D 2019 Nano Lett. 19 8928
Google Scholar
[37] Mills A, Hunte S L 1997 J. Photochem. Photobiol. A 108 1
Google Scholar
[38] Yang J, Hu Y, Jin C, Zhuge L, Wu X 2017 Thin Solid Films 637 9
Google Scholar
[39] Pan L, Xiao Y, Kuang X Y, Ju M 2021 Mater. Chem. Phys. 257 123824
Google Scholar
[40] Savin A, Nesper R, Wengert S, Fässler T F 1997 Angew. Chem. Int. Ed. Engl. 36 1808
Google Scholar
[41] Lu T, Chen F 2011 Acta Phys. Chim. Sin. 27 2786
Google Scholar
[42] Fuentealba P, Chamorro E, Santos J C 2007 Theoretical Aspects of Chemical Reactivity 19 57



 下载:
下载:
计量
- 文章访问数: 9478
- PDF下载量: 171
- 被引次数: 0
