










-
LiCoO2作为商业化最早的锂离子电池正极材料, 至今仍受到许多研究人员的广泛关注. 高电压下LiCoO2面临着严重的容量衰减和性能下降等问题, 实验上通常采用体相元素掺杂以稳定LiCoO2在高电压下的晶体结构, 从而提高其电化学性能. Mg元素掺杂被认为是一种能够提高LiCoO2高电压循环稳定性的有效手段, 但Mg的具体掺杂形式以及作用机理仍需进一步深入研究. 本文基于密度泛函理论的第一性原理计算研究了LiCoO2中Mg对Co位和Li位各种替代组态的形成能及其电子结构. 计算结果表明, Mg在LiCoO2中的替代情况较为复杂: 掺杂浓度为3.7%时, Mg更倾向于替代Co位; 而掺杂浓度提高至7.4%后, 则Mg不仅仅可以只替代Co位或Li位, 还存在同时替代Co位和Li位的可能; 各种替代组态也呈现出不同的电子态, 既存在金属态, 也有半导体态, 同时在许多情况下还伴有电子局域态. 因此, 我们认为LiCoO2的Mg掺杂位形与掺杂量有密切的关系, 且掺杂诱导的电子结构也存在较大的差异.Developing the cathode material with high voltage and high capacity is of critical importance in improving the energy density of the battery. Among various cathode materials, LiCoO2, as the first commercialized cathode material for lithium-ion batteries, is still widely concerned by many researchers due to its high output voltage, high volumetric energy density, and excellent cycling performance. However, a series of issues, such as serious capacity fading and performance deterioration, can emerge as cut-off voltage is above 4.5 V. Many strategies have been proposed to stabilize the cycling performance of LiCoO2 at high voltages. Mg doping is considered to be an effective strategy to improve the high voltage cycling stability of LiCoO2 cathode material, but the specific doping form and mechanism of Mg doping still need to be further studied. In this paper, the values of formation energy and the electronic structures of various configurations for Mg doping on Co and Li sites in LiCoO2 are investigated by the first-principles method based on density-functional theory. The calculated results show that the values of formation energy for different doping configurations are different and the substitution of Mg in LiCoO2 is complicated. When the doping concentration is 3.7%, Mg prefers to substitute for the Co site; while the doping concentration increases to 7.4%, Mg can replace not only the Co or Li sites, but also the Co and Li sites simultaneously. Therefore, it should not be simply believed that Mg ion can replace only Co or Li site in LiCoO2, depending on the specific doping situation actually. Furthermore, various doping configurations also exhibit different electronic states, including metallic state and semiconductor state, and what is more, electronic local states in many cases. Therefore, we believe that the Mg doping configuration in LiCoO2 is related closely to the doping amount, and the doping induced electronic structure also has a great difference.
-
Keywords:
- LiCoO2 /
- formation energy /
- electronic structure /
- first principles
[1] Li W D, Erickson E M, Manthiram A 2020 Nat. Energy 5 26
 Google Scholar
Google Scholar
[2] Yang X R, Lin M, Zheng G R, et al. 2020 Adv. Funct. Mater. 30 2004664
Google Scholar
[3] Aurbach D, Markovsky B, Rodkin A, Levi E, Cohen Y S, Kim H J, Schmidt M 2002 Electrochim. Acta 47 4291
Google Scholar
[4] Reddy M V, Jie T W, Jafta C J, et al. 2014 Electrochim. Acta 128 192
Google Scholar
[5] Lala S M, Montoro L A, Lemos V, Abbate M, Rosolen J M 2005 Electrochim. Acta 51 7
Google Scholar
[6] Abbate M, Lala S M, Montoro L A, Rosolen J M 2004 Phys. Rev. B 70 235101
Google Scholar
[7] 杨萧, 倪江锋, 黄友元, 陈继涛, 周恒辉, 张新祥 2006 物理化学学报 22 183
Google Scholar
Yang X, Ni J F, Huang Y Y, Chen J T, Zhou H H, Zhang X X 2006 Acta Phys. Chim. Sin. 22 183
Google Scholar
[8] Yu J P, Han Z H, Hu X H, Zhan H, Zhou Y H, Liu X J 2014 J. Power Sources 262 136
Google Scholar
[9] Wu K, Li Q, Chen M M, Chen D F, Wu M M, Hu Z B, Li F Q, Xiao X L 2018 J. Solid State Electrochem. 22 3725
Google Scholar
[10] Myung S T, Kumagai N, Komaba S, Chung H T 2001 Solid State Ionics 139 47
Google Scholar
[11] Xie M, Hu T, Yang L, Zhou Y 2016 RSC Adv. 6 63250
Google Scholar
[12] Julien C, Camacho-Lopez M A, Lemal M, Ziolkiewicz S 2002 Mater. Sci. Eng. B 95 6
Google Scholar
[13] Jin Y H, Xu S G. Li Z T, Xu K H, Ding W X, Song J W, Wang H B, Zhao J Q 2018 J. Electrochem. Soc. 165 A2267
Google Scholar
[14] Cheng T, Ma Z T, Qian R C, Wang Y T, Cheng Q, Lyu Y C, Nie A, Guo B K 2021 Adv. Funct. Mater. 31 2001974
Google Scholar
[15] Tian T, Zhang T W, Yin Y C, Tan Y H, Song Y H, Lu L L, Yao H B 2020 Nano Lett. 20 677
Google Scholar
[16] Zhang J N, Li Q H, Ouyang C Y, et al. 2019 Nat. Energy 4 594
Google Scholar
[17] Wang Z G, Wang Z X, Guo H J, Peng W J, Li X H 2015 Ceram. Int. 41 469
Google Scholar
[18] Tukamoto H, West A R 1997 J. Electrochem. Soc. 144 3164
Google Scholar
[19] Shi S Q, Ouyang C Y, Lei M S, Tang W H 2007 J. Power Sources 171 908
Google Scholar
[20] 徐晓光, 魏英进, 孟醒, 王春忠, 黄祖飞, 陈岗 2004 物理学报 53 210
Google Scholar
Xu X G, Wei Y J, Meng X, Wang C Z, Huang Z F, Chen G 2004 Acta Phys. Sin. 53 210
Google Scholar
[21] Varanasi A K, Bhowmik A, Sarkar T, Waghmare U V, Bharadwaj M D 2014 Ionics 20 315
Google Scholar
[22] Koyama Y, Arai H, Tanaka I, Uchimoto Y, Ogumi Z 2014 J. Mater. Chem. A 2 11235
Google Scholar
[23] Kresse G, Furthmüller J 1996 Comput. Mater. Sci. 6 15
Google Scholar
[24] Anisimov V I, Zaanen J, Andersen O K 1991 Phys. Rev. B 44 943
Google Scholar
[25] Ning F H, Li S, Xu B, Ouyang C Y 2014 Solid State Ionics 263 46
Google Scholar
[26] Zhou F, Cococcioni M, Marianetti C A, Morgan D, Ceder G 2004 Phys. Rev. B 70 235121
Google Scholar
[27] Monkhorst H J, Pack J D 1976 Phys. Rev. B 13 5188
Google Scholar
[28] Xu G G, Zhong K H, Yang Y M, Zhang J M, Huang Z G 2019 Solid State Ionics 338 25
Google Scholar
[29] Ning F H, Gong X, Rao F Y, Zeng X M, Ouyang C Y 2016 Int. J. Electrochem. Sci. 11 1951
[30] Chen Y, Yu Q, Xu G G, et al. 2019 ACS Appl. Mater. Interfaces 11 33043
Google Scholar
[31] Hoang K 2017 Phys. Rev. Mater. 1 075403
Google Scholar
-
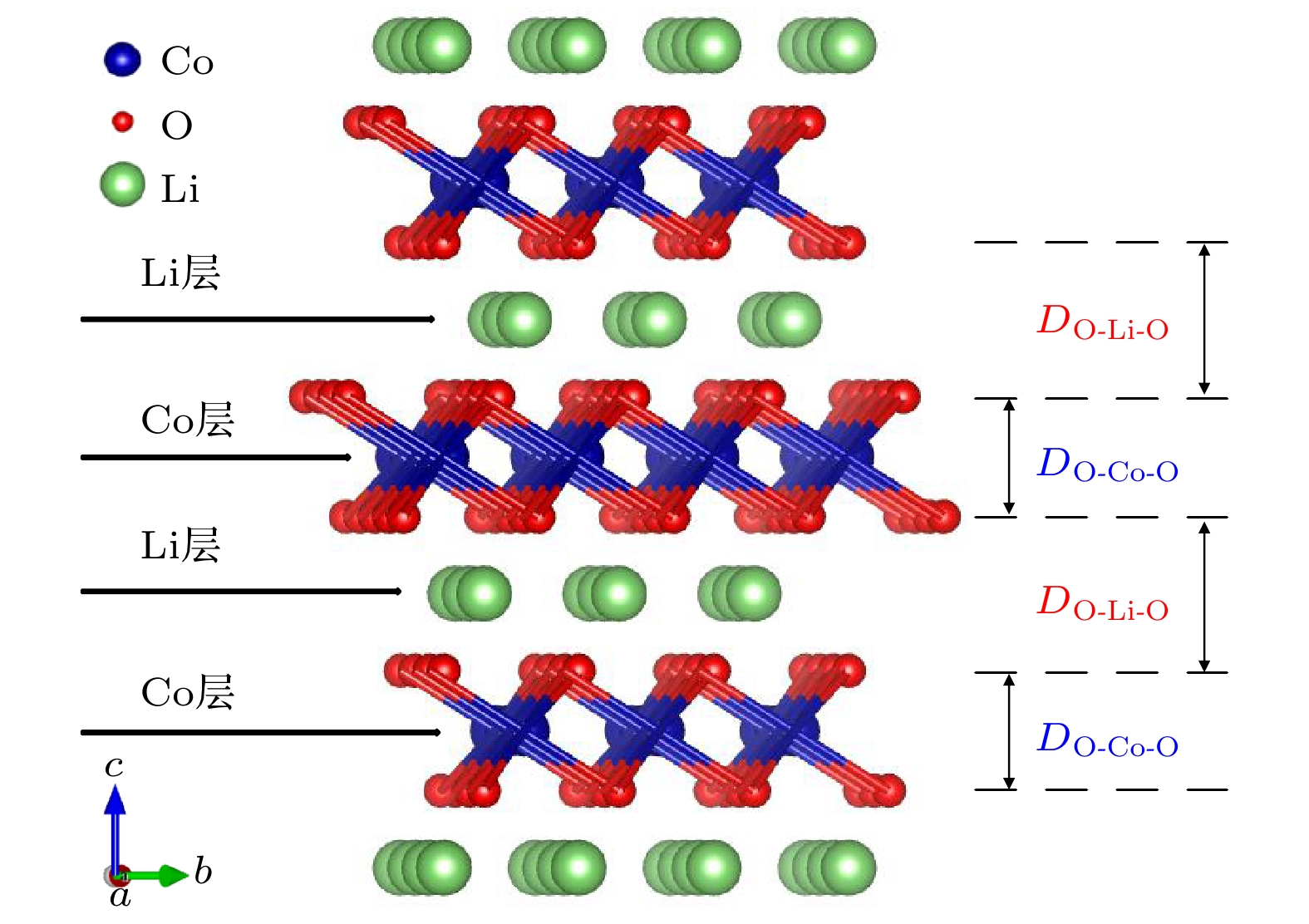
图 1 LiCoO2的晶胞结构, 符号“DO-Co-O”和“DO-Li-O”分别表示过渡金属层(Co层)和锂层(Li层)的厚度
Fig. 1. Unit cell of LiCoO2, the symbols of “DO-Co-O” and “DO-Li-O” are the oxygen distance across the transition metal layer (cobalt layer) and across the Li layer respectively.
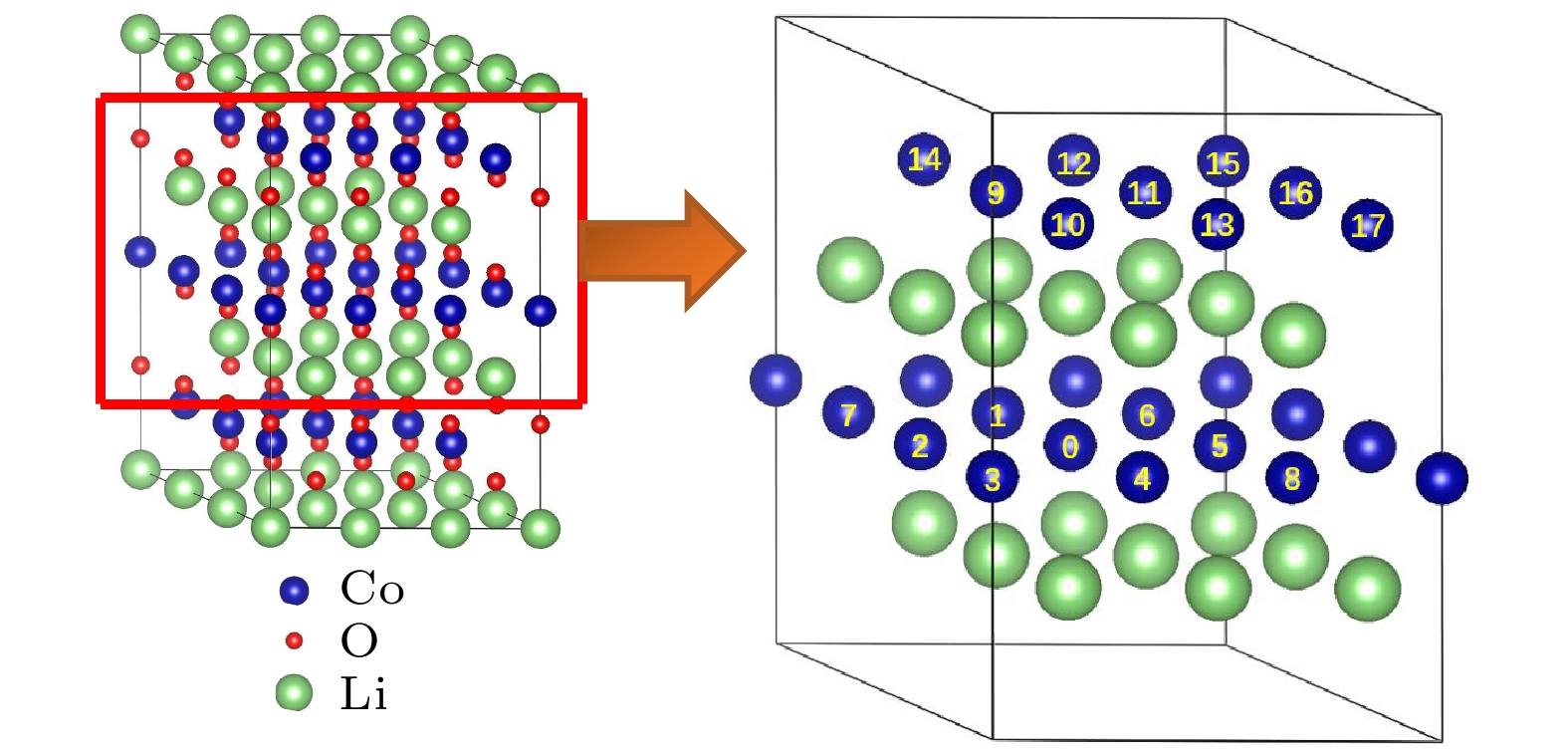
图 2 两个Mg替代两个Co位的各种组态示意图
Fig. 2. Schematic illustration of various configurations for two Mg replacing two Co sites.
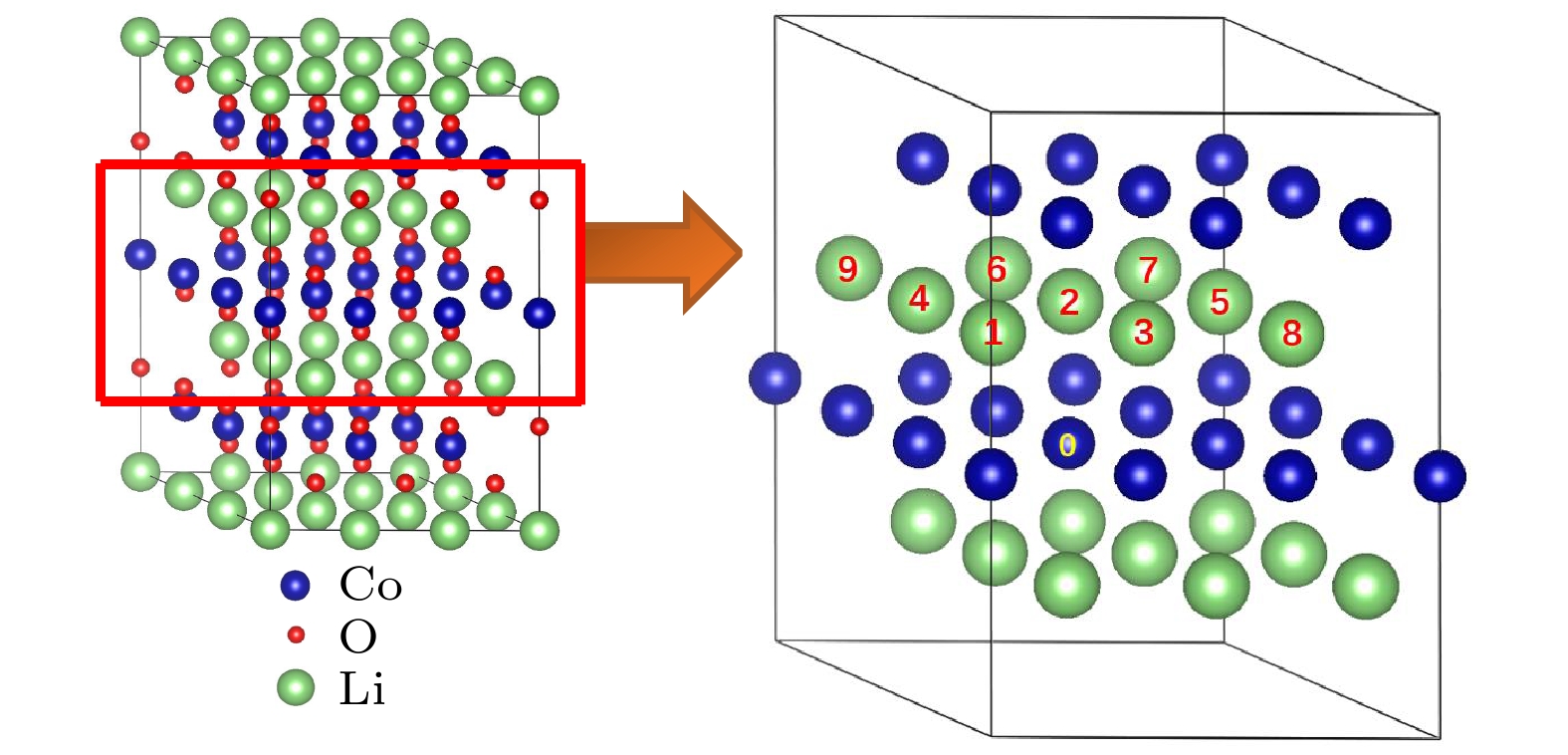
图 3 两个Mg分别替代1个Co位和1个Li位的各种组态示意图
Fig. 3. Schematic illustration of various configurations for two Mg replacing one Co site and oneLi site respectively.
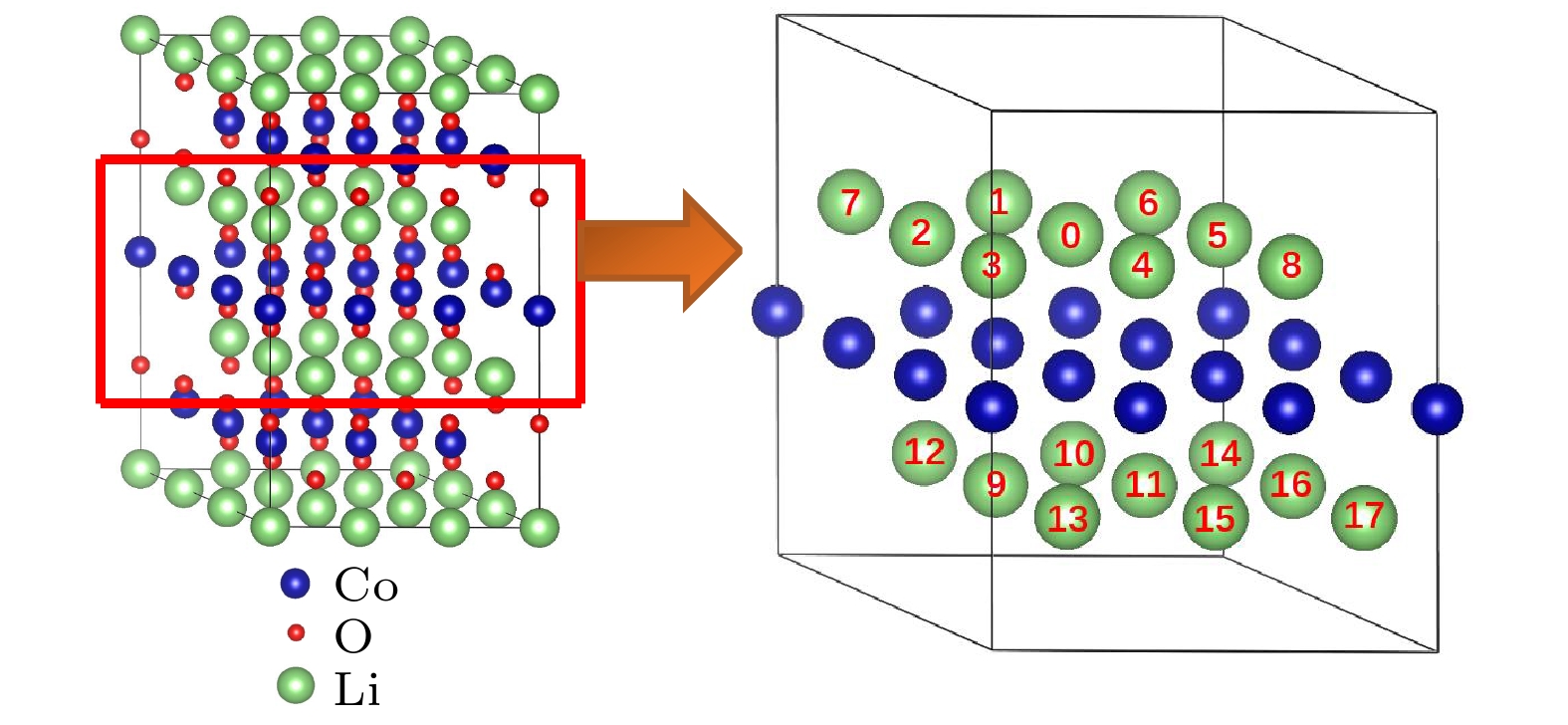
图 4 两个Mg替代两个Li位的各种组态示意图
Fig. 4. Schematic illustration of various configurations for two Mg replacing two Li sites.
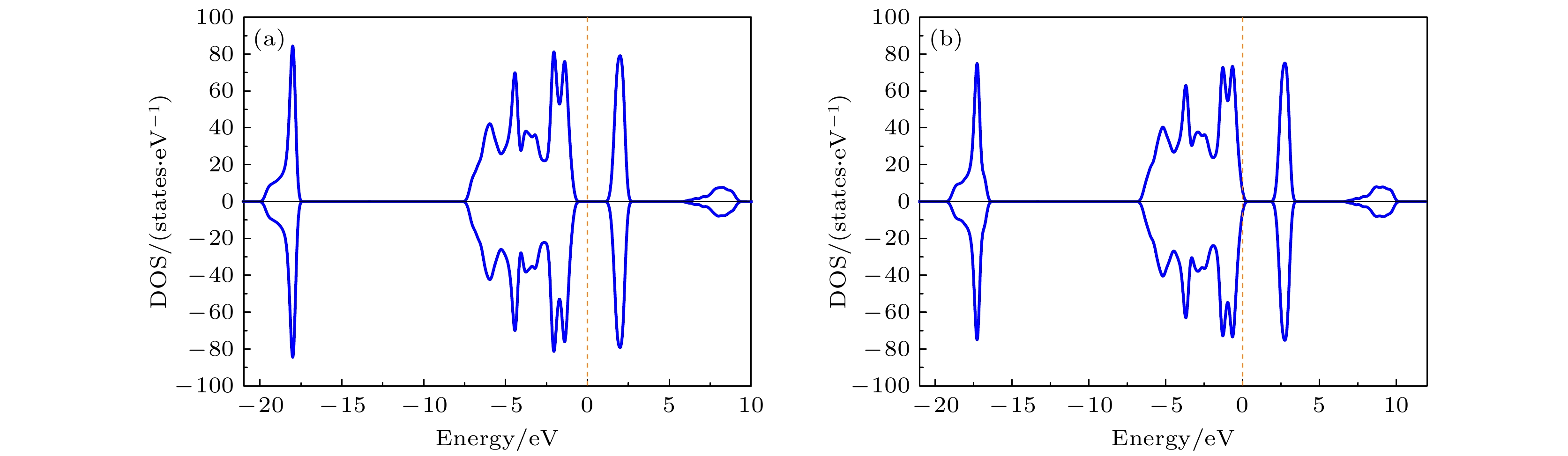
图 5 态密度(DOS)图 (a) 纯的LiCoO2; (b) LiCo0.963Mg0.037O2. 费米能级设为零
Fig. 5. Density of states (DOS): (a) Pure LiCoO2; (b) LiCo0.963Mg0.037O2. The Fermi level is set to be zero.
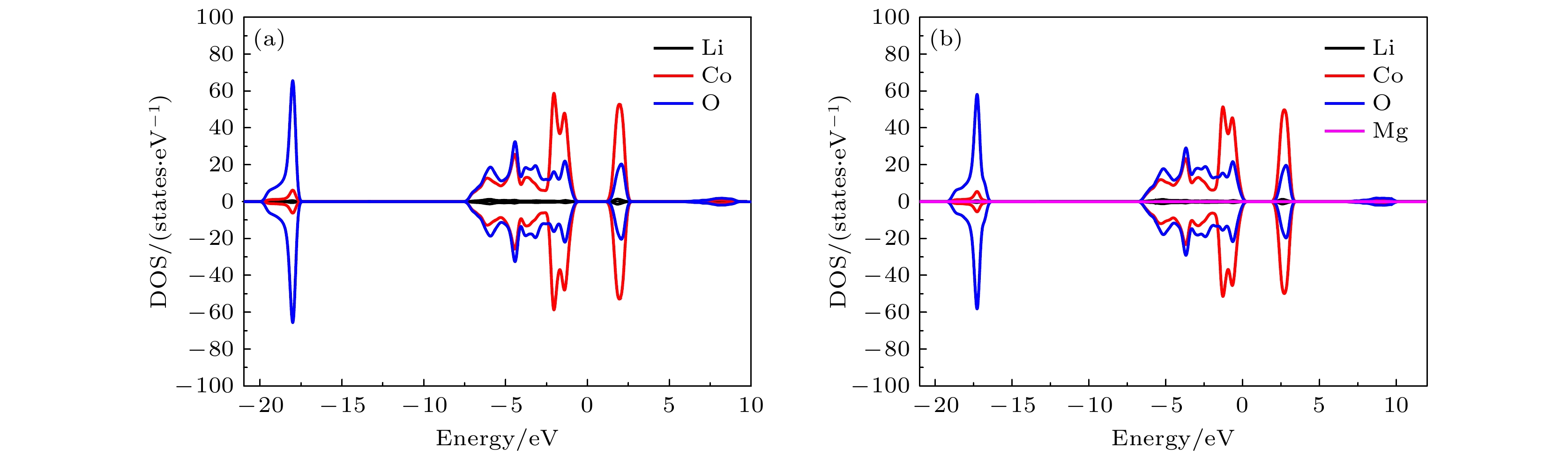
图 6 分波态密度(PDOS)图 (a) 纯的LiCoO2; (b) LiCo0.963Mg0.037O2
Fig. 6. Partial density of states (PDOS): (a) Pure LiCoO2; (b) LiCo0.963Mg0.037O2.
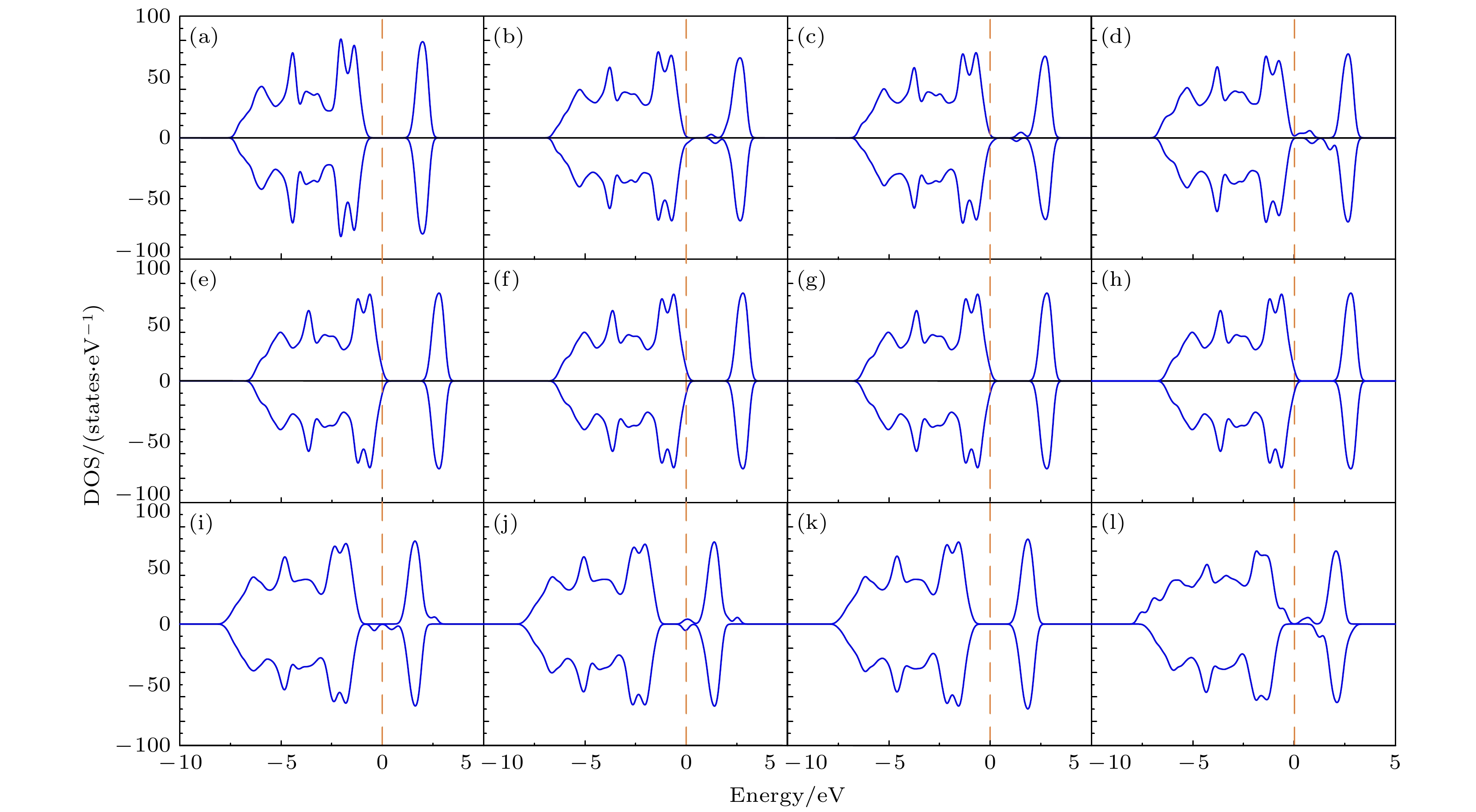
图 7 态密度(DOS)图 (a) 纯的LiCoO2; 图2所示的两个Mg替代同层的两个Co位时对应组态 (b) (0, 3), (c) (0, 5), (d) (0, 8); 图2所示的两个Mg替代异层的两个Co位时对应组态 (e) (0, 9), (f) (0, 12), (g) (0, 14), (h) (0, 17); 图3所示的两个Mg分别替代1个Co位和1个Li位时对应组态 (i) (0, 1), (j) (0, 2), (k) (0, 3), (l) (0, 5). 费米能级设为零
Fig. 7. Density of states (DOS): (a) Pure LiCoO2; (b) (0, 3), (c) (0, 5), (d) (0, 8)configurations for two Mg atoms replacing two Co sites in the same layer given in Fig. 2; (e) (0, 9), (f) (0, 12), (g) (0, 14), (h) (0, 17) configurations for two Mg atoms replacing two Co sites in different layers given in Fig. 2; (i) (0, 1), (j) (0, 2), (k) (0, 3), (l) (0, 5) configurations for two Mg atoms replacing one Co site and one Li site respectively given in Fig. 3. The Fermi level is set to be zero.

图 8 态密度(DOS)图. 图4中所示的两个Mg替代同层的两个Li位时对应组态 (a) (0, 1), (b) (0, 2), (c) (0, 3), (d)(0, 8); 图4中所示的两个Mg替代异层的两个Li位时对应组态 (e) (0, 10), (f) (0, 17). 费米能级设为零
Fig. 8. Density of states (DOS): (a) (0, 1), (b) (0, 2), (c) (0, 3), (d) (0, 8) configurations for two Mg atoms replacing two Li sites in the same layer given in Fig. 4; (e) (0, 10), (f) (0, 17) configurations for two Mg atoms replacing two Li sites in different layers given in Fig. 4. The Fermi level is set to be zero.
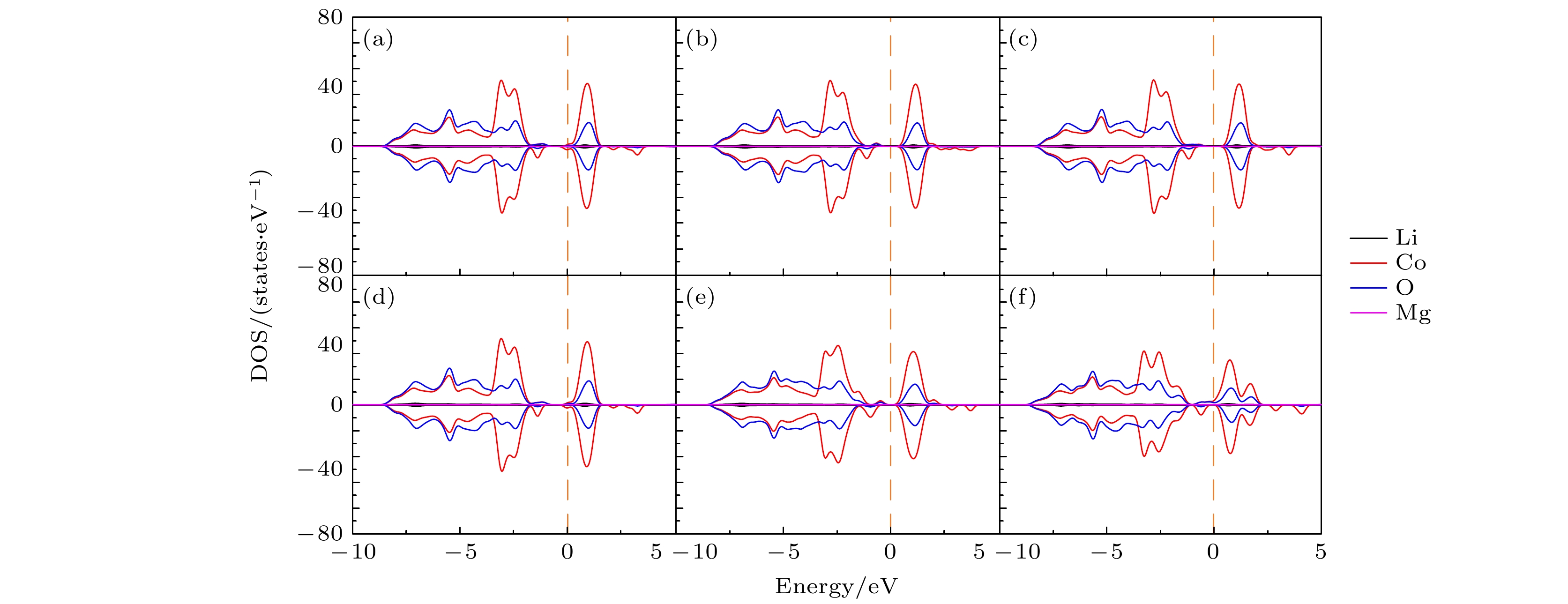
表 1 图2所示的各种掺杂组态的形成能
Table 1. Formation energies of the various doping configurations given in Fig. 2.
Mg-Mg间距d/Å 组态 Eform/eV 2.830 (0, 1) 1.651 (0, 2) 0.196 (0, 3) –0.588 (0, 4) 0.497 (0, 5) –0.604 (0, 6) 0.383 4.902 (0, 7) 0.348 (0, 8) –1.004 4.992 (0, 9) –0.887 (0, 10) –0.886 (0, 11) –0.886 5.738 (0, 12) –0.875 (0, 13) –0.874 6.398 (0, 14) –0.872 (0, 15) –0.870 (0, 16) –0.871 7.547 (0, 17) –0.655  下载: 导出CSV
下载: 导出CSV
表 2 图3所示的各种掺杂组态的形成能
Table 2. Formation energies of the various doping configurations given in Fig.3.
Mg-Mg间距d/Å 组态 Eform/eV 2.869 (0, 1) –1.137 (0, 2) –0.497 (0, 3) –2.228 4.030 (0, 4) 0.978 (0, 5) –0.143 4.924 (0, 6) 0.319 (0, 7) 0.346 (0, 8) 0.344 6.346 (0, 9) 0.756
下载: 导出CSV
表 3 图4所示的各种掺杂组态的形成能
Table 3. Formation energies of the various doping configurations given in Fig.4.
Mg-Mg间距d/Å 组态 Eform/eV 2.830 (0, 1) –0.965 (0, 2) –0.442 (0, 3) –0.635 (0, 4) –0.491 (0, 5) –0.723 (0, 6) –0.965 4.902 (0, 7) –0.126 (0, 8) –0.226 4.992 (0, 9) 1.883 (0, 10) –0.019 (0, 11) 0.208 5.738 (0, 12) 0.459 (0, 13) 0.729 (0, 14) 0.223 6.398 (0, 15) 2.305 (0, 16) 0.993 8.060 (0, 17) –0.182
下载: 导出CSV
-
[1] Li W D, Erickson E M, Manthiram A 2020 Nat. Energy 5 26
Google Scholar
[2] Yang X R, Lin M, Zheng G R, et al. 2020 Adv. Funct. Mater. 30 2004664
Google Scholar
[3] Aurbach D, Markovsky B, Rodkin A, Levi E, Cohen Y S, Kim H J, Schmidt M 2002 Electrochim. Acta 47 4291
Google Scholar
[4] Reddy M V, Jie T W, Jafta C J, et al. 2014 Electrochim. Acta 128 192
Google Scholar
[5] Lala S M, Montoro L A, Lemos V, Abbate M, Rosolen J M 2005 Electrochim. Acta 51 7
Google Scholar
[6] Abbate M, Lala S M, Montoro L A, Rosolen J M 2004 Phys. Rev. B 70 235101
Google Scholar
[7] 杨萧, 倪江锋, 黄友元, 陈继涛, 周恒辉, 张新祥 2006 物理化学学报 22 183
Google Scholar
Yang X, Ni J F, Huang Y Y, Chen J T, Zhou H H, Zhang X X 2006 Acta Phys. Chim. Sin. 22 183
Google Scholar
[8] Yu J P, Han Z H, Hu X H, Zhan H, Zhou Y H, Liu X J 2014 J. Power Sources 262 136
Google Scholar
[9] Wu K, Li Q, Chen M M, Chen D F, Wu M M, Hu Z B, Li F Q, Xiao X L 2018 J. Solid State Electrochem. 22 3725
Google Scholar
[10] Myung S T, Kumagai N, Komaba S, Chung H T 2001 Solid State Ionics 139 47
Google Scholar
[11] Xie M, Hu T, Yang L, Zhou Y 2016 RSC Adv. 6 63250
Google Scholar
[12] Julien C, Camacho-Lopez M A, Lemal M, Ziolkiewicz S 2002 Mater. Sci. Eng. B 95 6
Google Scholar
[13] Jin Y H, Xu S G. Li Z T, Xu K H, Ding W X, Song J W, Wang H B, Zhao J Q 2018 J. Electrochem. Soc. 165 A2267
Google Scholar
[14] Cheng T, Ma Z T, Qian R C, Wang Y T, Cheng Q, Lyu Y C, Nie A, Guo B K 2021 Adv. Funct. Mater. 31 2001974
Google Scholar
[15] Tian T, Zhang T W, Yin Y C, Tan Y H, Song Y H, Lu L L, Yao H B 2020 Nano Lett. 20 677
Google Scholar
[16] Zhang J N, Li Q H, Ouyang C Y, et al. 2019 Nat. Energy 4 594
Google Scholar
[17] Wang Z G, Wang Z X, Guo H J, Peng W J, Li X H 2015 Ceram. Int. 41 469
Google Scholar
[18] Tukamoto H, West A R 1997 J. Electrochem. Soc. 144 3164
Google Scholar
[19] Shi S Q, Ouyang C Y, Lei M S, Tang W H 2007 J. Power Sources 171 908
Google Scholar
[20] 徐晓光, 魏英进, 孟醒, 王春忠, 黄祖飞, 陈岗 2004 物理学报 53 210
Google Scholar
Xu X G, Wei Y J, Meng X, Wang C Z, Huang Z F, Chen G 2004 Acta Phys. Sin. 53 210
Google Scholar
[21] Varanasi A K, Bhowmik A, Sarkar T, Waghmare U V, Bharadwaj M D 2014 Ionics 20 315
Google Scholar
[22] Koyama Y, Arai H, Tanaka I, Uchimoto Y, Ogumi Z 2014 J. Mater. Chem. A 2 11235
Google Scholar
[23] Kresse G, Furthmüller J 1996 Comput. Mater. Sci. 6 15
Google Scholar
[24] Anisimov V I, Zaanen J, Andersen O K 1991 Phys. Rev. B 44 943
Google Scholar
[25] Ning F H, Li S, Xu B, Ouyang C Y 2014 Solid State Ionics 263 46
Google Scholar
[26] Zhou F, Cococcioni M, Marianetti C A, Morgan D, Ceder G 2004 Phys. Rev. B 70 235121
Google Scholar
[27] Monkhorst H J, Pack J D 1976 Phys. Rev. B 13 5188
Google Scholar
[28] Xu G G, Zhong K H, Yang Y M, Zhang J M, Huang Z G 2019 Solid State Ionics 338 25
Google Scholar
[29] Ning F H, Gong X, Rao F Y, Zeng X M, Ouyang C Y 2016 Int. J. Electrochem. Sci. 11 1951
[30] Chen Y, Yu Q, Xu G G, et al. 2019 ACS Appl. Mater. Interfaces 11 33043
Google Scholar
[31] Hoang K 2017 Phys. Rev. Mater. 1 075403
Google Scholar

 下载:
下载:
计量
- 文章访问数: 13492
- PDF下载量: 393
- 被引次数: 0
