










-
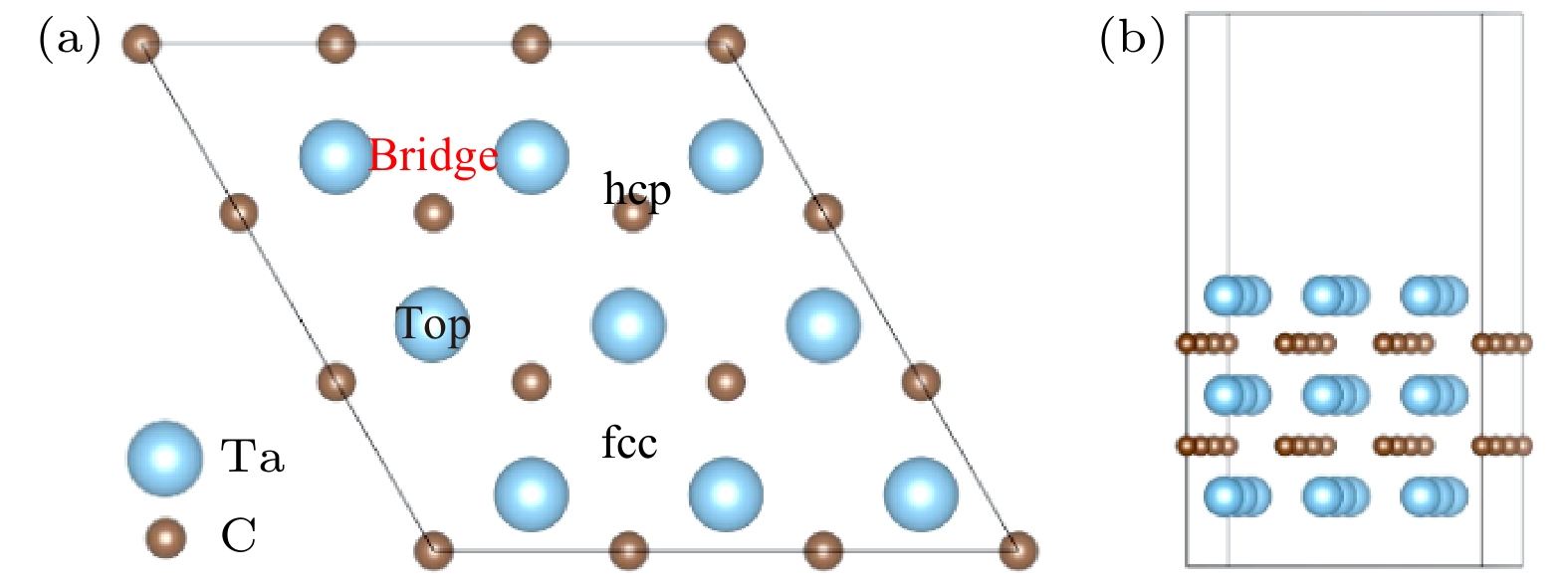
采用自旋极化密度泛函理论(DFT)并结合周期平板模型的方法, 研究了NH3在TaC表面的吸附和分解反应机理. 表面能计算结果显示, 以Ta为终止的TaC(0001)面为最稳定的表面; NH3分子通过其孤对电子优先吸附在顶位top位, 而NH2和H最稳定吸附位置为三重hcp位, NH和N吸附在三重fcc位. 过渡态结果表明氮原子的复合反应脱附为整个反应的限速步骤. 电子结构计算结果表明, NH3分子及其片段通过其N原子的2pz轨道与底物Ta的5
$ {\rm d}_{z^2} $ 轨道混合吸附于表面. 随着脱氢反应的进行, 电荷转移现象变得逐渐明显, 吸附质和底物之间的电荷转移在加速NH3脱氢催化过程中发挥重要作用.The adsorption and desorption behaviors of ammonia on TaC(0001) surface are studied by employing spin-polarized density function theory calculations. The surface energy calculation results show that the TaC (0001) terminating with Ta is the most stable surface. According to the optimized structural and energetic properties, it is found that NH3 prefers to adsorb on the top site, whereas NH2, H prefer to adsorb on the triple hcp site and NH, N prefer to stay on the triple fcc site. In addition, three transition states are found for analyzing the mechanism of dehydrogenation of NH3, and the N recombination reaction is also considered. The results show that the desorption of nitrogen atoms is the rate-determining step in the overall reaction. Finally, in order to further elucidate the mechanism of NH3 adsorption and dissociation on the surface of Ta-TaC, the electronic structure of the most stable adsorption position is analyzed from the perspective of charge density distribution and electron density of states. The results of electronic structure calculation show that NH3 molecule is adsorbed on the surface through the mixture of 2pz orbital of N atom and$ 5{\rm d}_{z^2} $ orbital of substrate Ta. With the progress of dehydrogenation, the charge transfer phenomenon becomes more and more serious. The charge transfer between adsorbate and substrate plays an important role in accelerating NH3 dehydrogenation catalytic process.-
Keywords:
- density functional theory /
- ammonia /
- TaC /
- adsorption
[1] Hansgen D A, Vlachos D G, Chen J G G 2010 Nat. Chem. 2 484
 Google Scholar
Google Scholar
[2] Lee Y J, Lee Y S, Cha J Y, et al. 2020 Int. J. Hydrogen Energy 45 19181
Google Scholar
[3] Kirste K G, McAulay K, Bell T E, et al. 2021 Appl. Catal. B 280 119405
Google Scholar
[4] Li L, Chu W, Ding C, Xi X G, Jiang R Y, Yan J L 2017 Int. J. Hydrogen Energy 42 30630
Google Scholar
[5] Cui X Z, Li H, Guo L M, He D N, Chen H, Shi J L 2008 Dalton Trans. 6435
[6] Pansare S S, Torres W, Goodwin J G 2007 Catal. Commun. 8 649
Google Scholar
[7] Choi J G 1999 J. Catal. 182 104
Google Scholar
[8] Zheng W Q, Cotter T P, Kaghazchi P, et al. 2013 J. Am. Chem. Soc. 135 3458
Google Scholar
[9] Choi J G 2013 J. Porous Mater. 20 1059
Google Scholar
[10] Choi J G 1999 Appl. Catal. A 184 189
Google Scholar
[11] Yi W, Tang G, Chen X, Yang B, Liu X 2020 Comput. Phys. Commun. 257 107535
Google Scholar
[12] Hsiao M K, Su C H, Liu C Y, Chen H L 2015 Phys. Chem. Chem. Phys. 17 30598
Google Scholar
[13] 马淳安, 刘婷, 陈丽涛 2010 物理化学学报 26 155
Google Scholar
Ma C A, Liu T, Chen L T 2010 Acta Phys. -Chim. Sin. 26 155
Google Scholar
[14] Yeo S C, Han S S, Lee H M 2014 J. Phys. Chem. C 118 5309
Google Scholar
[15] Jiang Z, Qin P, Fang T 2016 Appl. Surf. Sci. 371 337
Google Scholar
[16] Hsiao M K, Wu S K, Chen H L 2015 J. Phys. Chem. C 119 4188
[17] Walkosz W, Zapol P, Stephenson G B 2012 J. Chem. Phys. 137 054708
Google Scholar
[18] Zhou S L, Lin S, Guo H 2018 J. Phys. Chem. C 122 9091
Google Scholar
[19] Jiang Z, Pan Q, Li M M, Yan T, Fang T 2014 Appl. Surf. Sci. 292 494
Google Scholar
[20] Pillai H S, Xin H L 2019 Ind. Eng. Chem. Res. 58 10819
Google Scholar
[21] 肖香珍, 杨理 2019 原子与分子物理学报 36 59
Xiao X Z, Yang L 2019 J. At. Mol. Phys. 36 59
[22] Tang S B, Cao Z X 2012 J. Phys. Chem. C 116 8778
Google Scholar
-
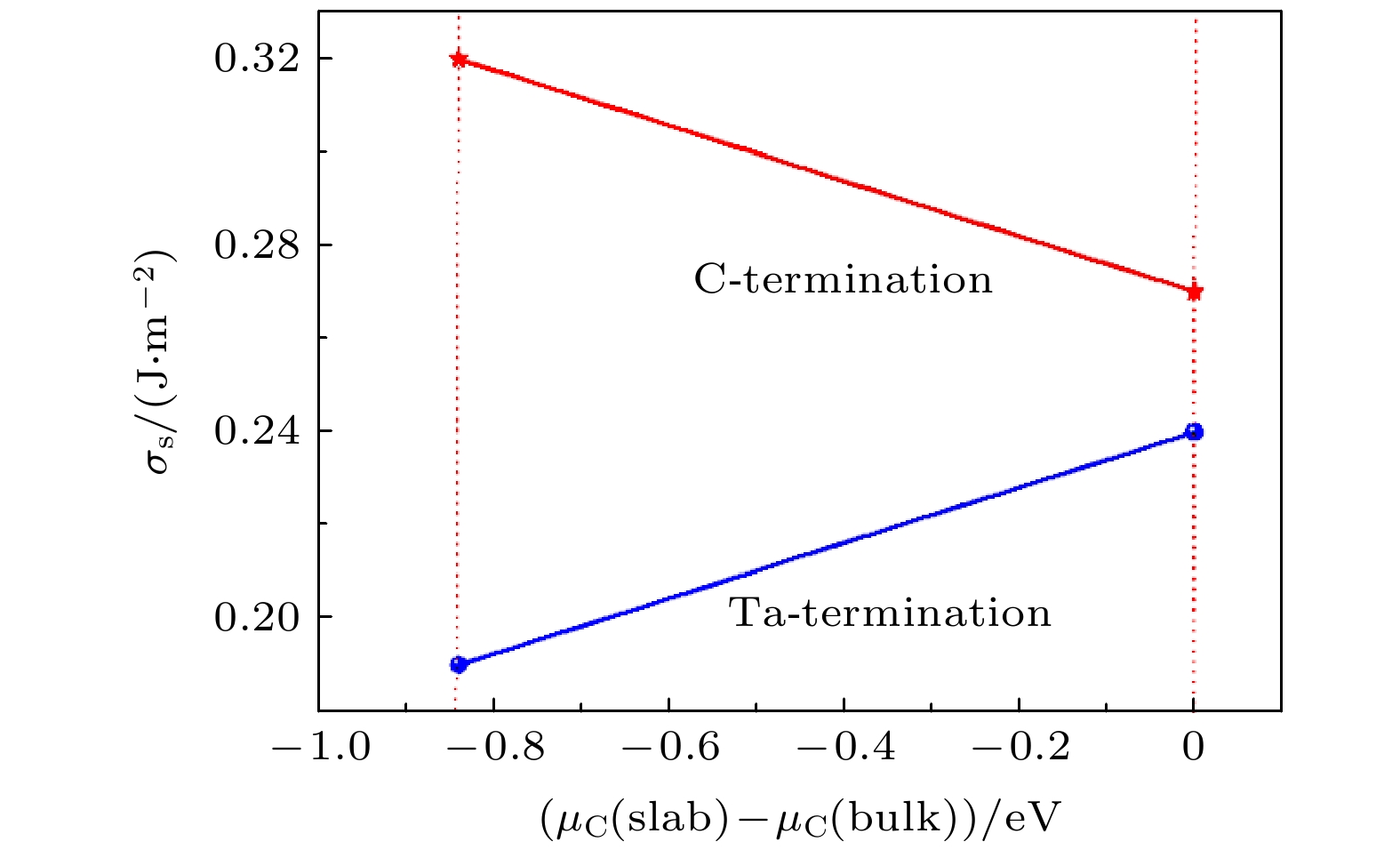
图 1 TaC(0001)的表面能(
$ {\sigma }_{\mathrm{s}} $ )与化学势之间的关系Fig. 1. Relationships between surface energies of two TaC (0001) surfaces and chemical potentials.
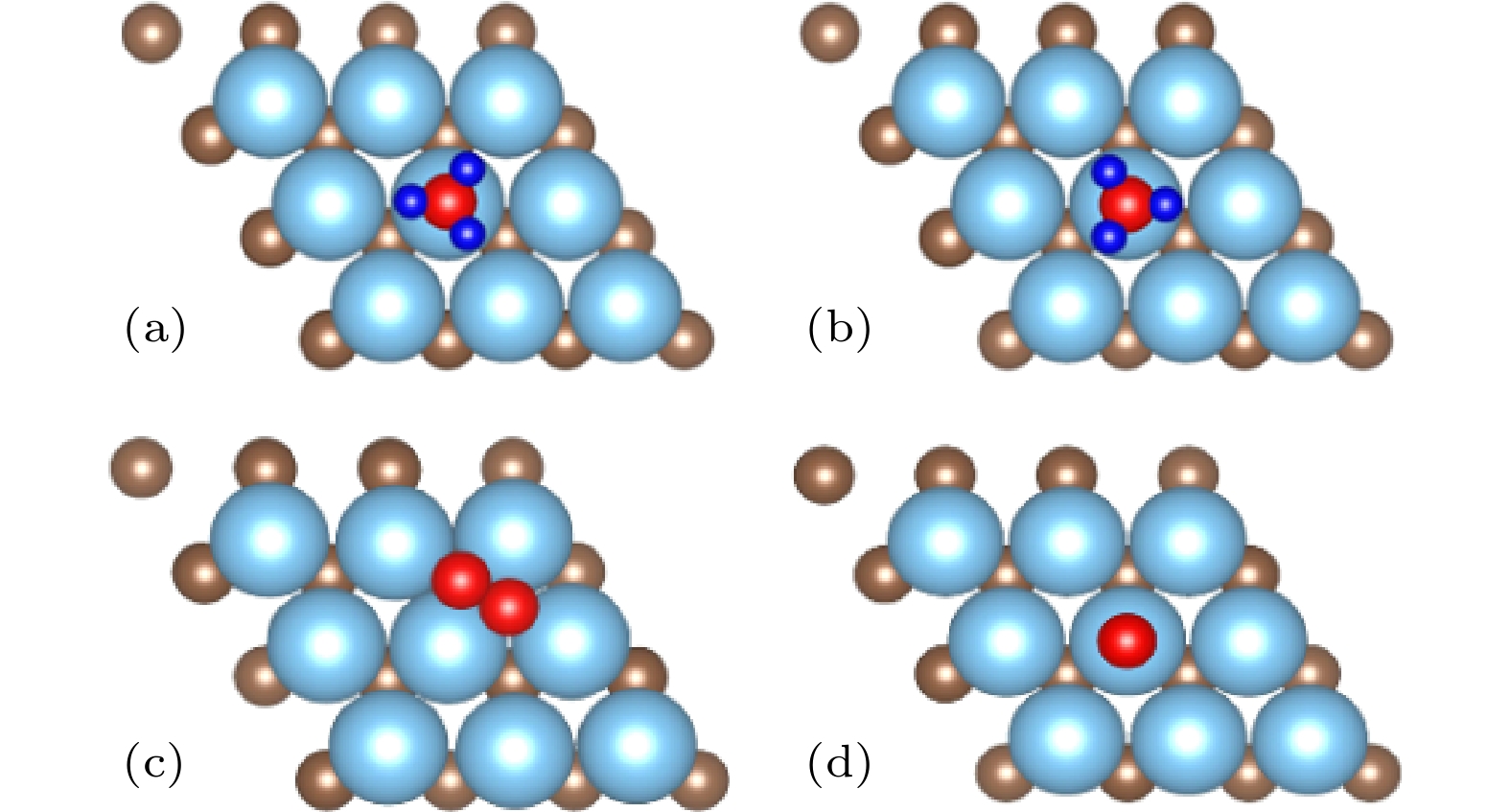
图 3 (a)(b) NH3和 (c)(d) N2的两种吸附构型
Fig. 3. Two adsorption configurations of (a)(b) NH3 and (c)(d) N2.

图 4 NH3脱氢和复合反应的初态、过渡态和终态结构
Fig. 4. Structure of initial state, transition state and final state of NH3 dehydrogenation and recombination reaction.
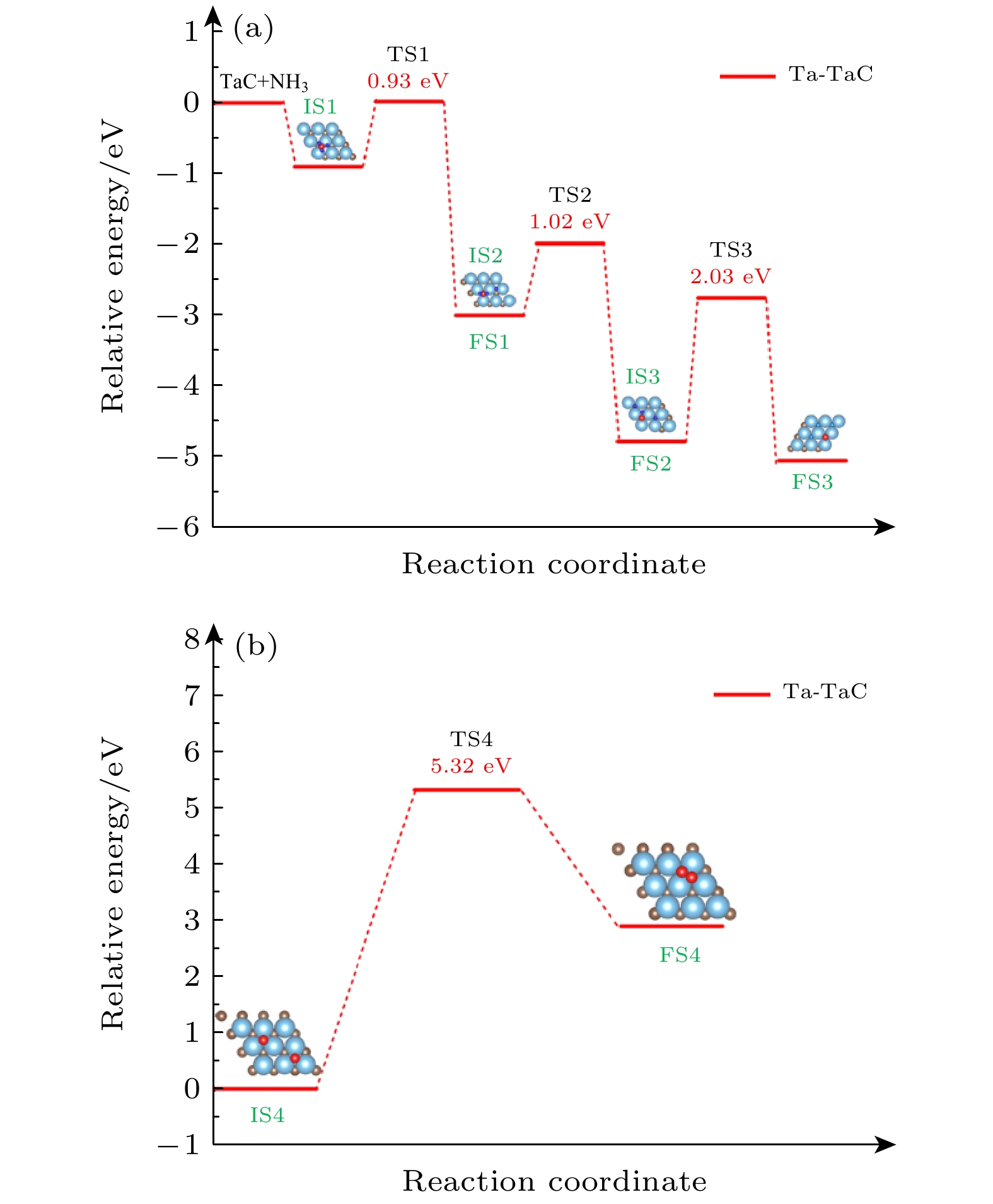
图 5 (a) NH3分解反应和 (b) N-N复合反应的能量图
Fig. 5. Calculated potential energy diagram for (a) NH3 dehydrogenation and (b) N-N coupling reaction.
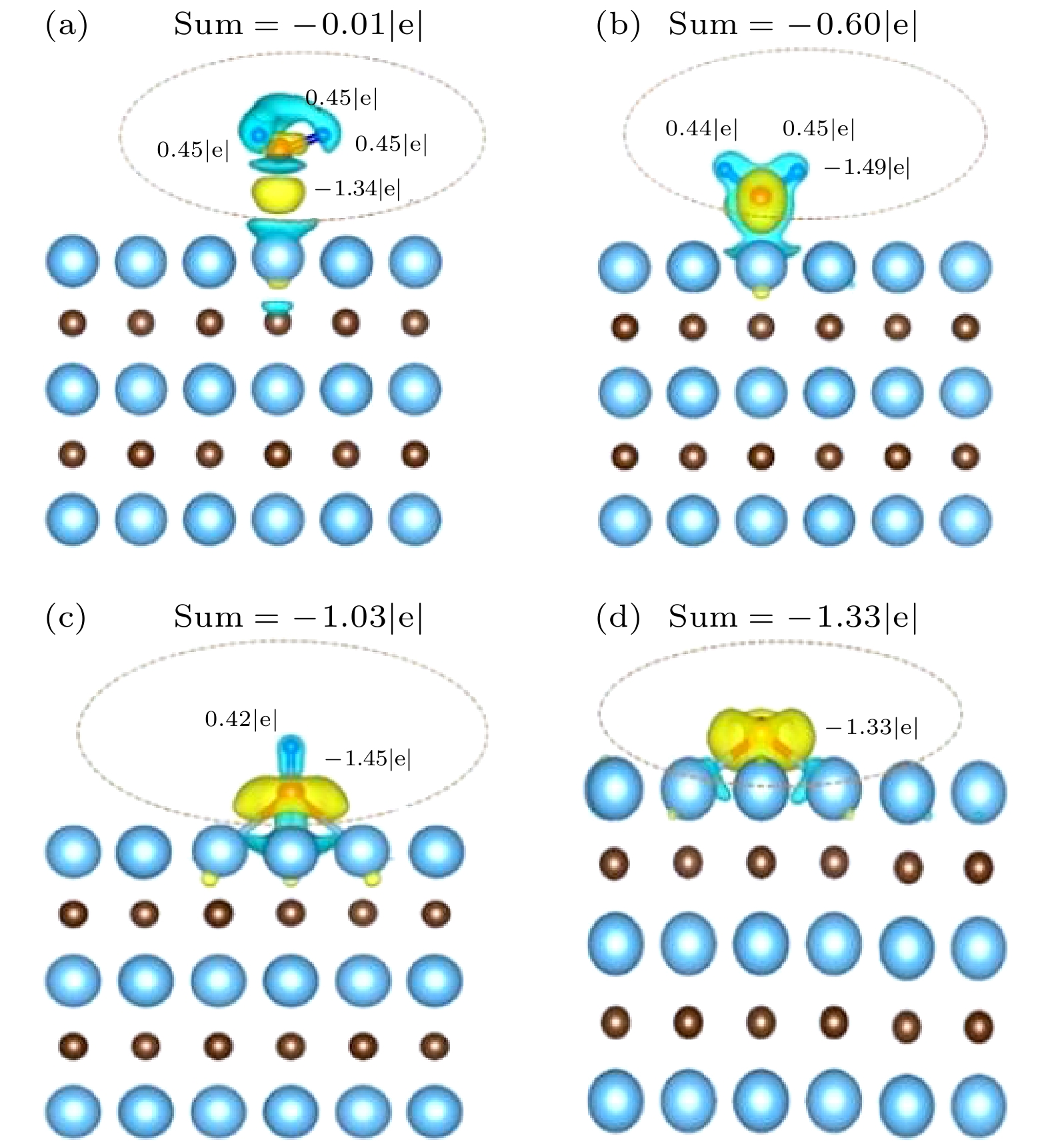
图 6 (a) NH3, (b) NH2, (c) NH, (d) N在Ta-TaC表面的差分电荷密度示意图和Bader电荷分析
Fig. 6. Schematic diagram of differential charge density of (a) NH3, (b) NH2, (c) NH, (d) N on the surface of Ta-TaC and Bader charge analysis.

图 7 (a) 吸附前NH3, (b) 吸附后NH3, (c) NH2, (d) NH, (e) N吸附于Ta-TaC表面态密度分布, 虚线代表费米能级
Fig. 7. Local density of states (LDOS) for NH3 dehydrogenation: (a) NH3 before interaction, (b) NH3 after interaction, (c) NH2, (d) NH, (e) N The dashed line represents the Fermi level.
表 1 Ta-TaC表面的吸附位点、吸附能和关键几何参数
Table 1. Adsorption site, adsorption energy and key geometric parameters of Ta-TaC surface.
Species Sites Eads/eV d (N—Ta) d (N—H) θ(H—N—H) NH3 top 0.08 2.354 1.027 108.1 NH2 hcp –3.6 2.221 1.036 102.8 NH fcc –6.49 2.140 1.026 — N fcc –9.79 2.030 — — H hcp –1.1 — — — v-N2 top –0.37 2.157 — —  下载: 导出CSV
下载: 导出CSV
表 2 各步基元反应的活化能、虚频和反应热
Table 2. The activation energy、imaginary frequency and reaction heat of each step elementary reaction.
reaction $ \Delta E $/eV $ \Delta H $/eV Freq./cm–1 $ {\rm{ N}}{{\text{H}}_3} \to {\rm{ N}}{{\text{H}}_{2}}+{\text{H}} $ 0.93 –2.10 1154 $ {\rm{ N}}{{\text{H}}_2} \to {\text{NH}}+{\text{H}} $ 1.02 –1.78 502 $ {\text{NH}} \to {\text{N}} + {\text{H}} $ 2.03 –0.27 1405 $ {\text{N}} + {\text{N}} \to {{\rm{ N}}_2} $ 5.32 2.89 136
下载: 导出CSV
-
[1] Hansgen D A, Vlachos D G, Chen J G G 2010 Nat. Chem. 2 484
Google Scholar
[2] Lee Y J, Lee Y S, Cha J Y, et al. 2020 Int. J. Hydrogen Energy 45 19181
Google Scholar
[3] Kirste K G, McAulay K, Bell T E, et al. 2021 Appl. Catal. B 280 119405
Google Scholar
[4] Li L, Chu W, Ding C, Xi X G, Jiang R Y, Yan J L 2017 Int. J. Hydrogen Energy 42 30630
Google Scholar
[5] Cui X Z, Li H, Guo L M, He D N, Chen H, Shi J L 2008 Dalton Trans. 6435
[6] Pansare S S, Torres W, Goodwin J G 2007 Catal. Commun. 8 649
Google Scholar
[7] Choi J G 1999 J. Catal. 182 104
Google Scholar
[8] Zheng W Q, Cotter T P, Kaghazchi P, et al. 2013 J. Am. Chem. Soc. 135 3458
Google Scholar
[9] Choi J G 2013 J. Porous Mater. 20 1059
Google Scholar
[10] Choi J G 1999 Appl. Catal. A 184 189
Google Scholar
[11] Yi W, Tang G, Chen X, Yang B, Liu X 2020 Comput. Phys. Commun. 257 107535
Google Scholar
[12] Hsiao M K, Su C H, Liu C Y, Chen H L 2015 Phys. Chem. Chem. Phys. 17 30598
Google Scholar
[13] 马淳安, 刘婷, 陈丽涛 2010 物理化学学报 26 155
Google Scholar
Ma C A, Liu T, Chen L T 2010 Acta Phys. -Chim. Sin. 26 155
Google Scholar
[14] Yeo S C, Han S S, Lee H M 2014 J. Phys. Chem. C 118 5309
Google Scholar
[15] Jiang Z, Qin P, Fang T 2016 Appl. Surf. Sci. 371 337
Google Scholar
[16] Hsiao M K, Wu S K, Chen H L 2015 J. Phys. Chem. C 119 4188
[17] Walkosz W, Zapol P, Stephenson G B 2012 J. Chem. Phys. 137 054708
Google Scholar
[18] Zhou S L, Lin S, Guo H 2018 J. Phys. Chem. C 122 9091
Google Scholar
[19] Jiang Z, Pan Q, Li M M, Yan T, Fang T 2014 Appl. Surf. Sci. 292 494
Google Scholar
[20] Pillai H S, Xin H L 2019 Ind. Eng. Chem. Res. 58 10819
Google Scholar
[21] 肖香珍, 杨理 2019 原子与分子物理学报 36 59
Xiao X Z, Yang L 2019 J. At. Mol. Phys. 36 59
[22] Tang S B, Cao Z X 2012 J. Phys. Chem. C 116 8778
Google Scholar




 下载:
下载:
计量
- 文章访问数: 11656
- PDF下载量: 290
- 被引次数: 0
