










-
采用经典分子动力学模拟, 结合第一性原理计算及晶格动力学方法对氮化镓(GaN)纤锌矿结构与岩盐结构在 0—80 GPa压力范围内的相图进行了预测. 第一性原理计算与分子动力学模拟得到的零温下 GaN纤锌矿到岩盐结构的相变压力分别为 44.3 GPa 和 45.9 GPa, 与已有研究的实验结果吻合; 通过外推纤锌矿结构 GaN的熔化曲线得到其零压下的熔化温度为 2295 K, 当压力增大到33.3 GPa 时, 纤锌矿结构熔化曲线与岩盐结构熔化曲线相交, 两种结构的熔化温度均随压力的增大而上升; GaN还可能存在超离子相, 纤锌矿结构在压力大于2.0 GPa 且温度高于 2550 K时发生超离子相转变, 岩盐结构在压力温度大于 33.1 GPa 和 4182 K 后发生超离子相转变, 二者的相转变温度均会随着压力的增大而升高; GaN纤锌矿和岩盐结构的相界线并非为一直线, 在高温下相界线斜率为正, 随着温度的降低逐渐变为一条具有负斜率的曲线.The III-V compound semiconductor, GaN, has become an excellent semiconductor material for developing the high-frequency and high-power electronic devices because of its excellent characteristics, including large band width, high thermal conductivity and fast electron saturation rate, and has received extensive attention in recent years. However, the decomposition temperature of GaN is lower than the melting temperature, some of its fundamental properties, such as melting temperature and high temperature phase transition pressure, are still unclear, and so, now the investigation of fundamental properties dominates the whole process of this material from development to mature applications. In the present work, the classical molecular dynamics simulations combined with the first-principles calculations and lattice dynamics methods are adopted to predict the phase diagrams of GaN with wurtzite and rocksalt structures in a pressure range of 0–80 GPa. The phase transition pressures, 44.3 GPa and 45.9 GPa, obtained from the first-principles calculations and molecular dynamics simulations from wurtzite to rocksalt structure in GaN at zero temperature, are in agreement with the available experimental results (Sadovyi B, et al.
2020 Phys. Rev. B 102 235109 ). The melting temperature at 0 GPa is 2295 K obtained by extrapolating the GaN melting curve of the wurtzite structure. With the pressure increasing to 33.3 GPa, the melting curve of wurtzite structure in GaN intersects with the melting curve of rocksalt structure, and the melting temperatures of both structures increase with pressure increasing. It is found that GaN may have a superionic phase and the superionic phase transition occurs in the wurtzite structure at pressures greater than 2.0 GPa and temperatures above 2550 K, whereas the rocksalt structure undergoes a superionic phase transition at pressures and temperatures higher than 33.1 GPa and 4182 K, respectively, and both of the phase transition temperatures increase with pressure increasing. The slope of the phase boundary line of GaN is positive at high temperatures and gradually changes into a curve with a negative slope as the temperature decreases.-
Keywords:
- GaN /
- Phase diagram /
- Melting temperature /
- Molecular dynamics simulations
[1] Abdul Amir H A A, Fakhri M A, Abdulkhaleq Alwahib A 2021 Mater. Today Proc. 42 2815
 Google Scholar
Google Scholar
[2] Van Vechten J A 1973 Phys. Rev. B 7 1479
Google Scholar
[3] Zhou Y, Wang S F, Wang R, Jiang N 2013 Physica B 431 115
Google Scholar
[4] Sadovyi B, Wierzbowska M, Stelmakh S, Boccato S, Gierlotka S, Irifune T, Porowski S, Grzegory I 2020 Phys. Rev. B 102 235109
Google Scholar
[5] Utsumi W, Saitoh H, Kaneko H, Watanuki T, Aoki K, Shimomura O 2003 Nat. Mater. 2 735
Google Scholar
[6] Harafuji K, Tsuchiya T, Kawamura K 2004 J. Appl. Phys. 96 2501
Google Scholar
[7] Karpiński J, Jun J, Porowski S 1984 J. Cryst. Growth 66 1
Google Scholar
[8] Xia H, Xia Q, Ruoff A L 1993 Phys. Rev. B 47 12925
Google Scholar
[9] Novikov S V, Zainal N, Akimov A V, Staddon C R, Kent A J, Foxon C T 2010 J. Vac. Sci. Technol. B 28 C3B1
Google Scholar
[10] Ueno M, Yoshida M, Onodera A, Shimomura O, Takemura K 1994 Phys. Rev. B 49 14
Google Scholar
[11] Saoud F S, Plenet J C, Louail L, Maouche D 2011 Comput. Theor. Chem. 964 65
Google Scholar
[12] Sokol A G, Palyanov Y N, Surovtsev N V 2007 Diam. Relat. Mater. 16 431
Google Scholar
[13] Saitoh H, Utsumi W, Kaneko H, Aoki K 2007 J. Cryst. Growth 300 26
Google Scholar
[14] Porowski S, Sadovyi B, Gierlotka S, Rzoska S J, Grzegory I, Petrusha I, Turkevich V, Stratiichuk D 2015 J. Phys. Chem. Solids 85 138
Google Scholar
[15] Nord J, Albe K, Erhart P, Nordlund K 2003 J. Phys. Condens. Matter 15 5649
Google Scholar
[16] Alder B J, Wainwright T E 1957 J. Chem. Phys. 27 1208
Google Scholar
[17] Kioseoglou J, Polatoglou H M, Lymperakis L, Nouet G, Komninou P 2003 Comp. Mater. Sci. 27 43
Google Scholar
[18] Béré A, Serra A 2006 Philos. Mag. 86 2159
Google Scholar
[19] Do E C, Shin Y H, Lee B J 2009 J. Phys. :Condens. Matter 21 325801
Google Scholar
[20] Zapol P, Pandey R, Gale J D 1997 J. Phys. :Condens. Matter 9 9517
Google Scholar
[21] Zhang S, Chen N X 2005 Chem. Phys. 309 309
Google Scholar
[22] Born M, Mayer J E 1932 Z. Physik 75 1
Google Scholar
[23] Fischer T H, Almlof J 1992 J. Phys. Chem. 96 9768
Google Scholar
[24] Perdew J P, Ruzsinszky A, Csonka G I, Vydrov O A, Scuseria G E, Constantin L A, Zhou X, Burke K 2008 Phys. Rev. Lett. 100 136406
Google Scholar
[25] Vanderbilt D 1990 Phys. Rev. B 41 7892
Google Scholar
[26] Clark S J, Segall M D, Pickard C J, Hasnip P J, Probert M I, Refson K, Payne M C 2005 Z. Kristallogr. 220 567
[27] Gale J D, Rohl A L 2003 Mol. Simulat. 29 291
Google Scholar
[28] Pandey R, Jaffe J E, Harrison N M 1994 J. Phys. Chem. Solids 55 1357
Google Scholar
[29] Kirchner V, Heinke H, Hommel D, Domagala J Z, Leszczynski M 2000 Appl. Phys. Lett. 77 1434
Google Scholar
[30] Adachi K, Ogi H, Nagakubo A, Nakamura N, Hirao M, Imade M, Yoshimura M, Mori Y 2016 J. Appl. Phys. 119 245111
Google Scholar
[31] Polian A, Grimsditch M, Grzegory I 1996 J. Appl. Phys. 79 3343
Google Scholar
[32] Plimpton S 1995 J. Comput. Phys. 117 1
Google Scholar
[33] Zou Y C, Xiang S K, Dai C D 2020 Comp. Mater. Sci. 171 109156
Google Scholar
[34] Hazarika M P, Chakraborty S N 2019 Chem. Phys. Lett. 730 521
Google Scholar
[35] Liu C M, Xu C, Cheng Y, Chen X R, Cai L C 2015 J. Appl. Phys. 118 235901
Google Scholar
[36] Wang S C, Zhang G M, Liu H F, Song H F 2013 J. Chem. Phys. 138 134101
Google Scholar
[37] Cazorla C, Errandonea D 2013 J. Phys. Chem. C 117 11292
[38] Porowski S, Sadovyi B, Karbovnyk I, Gierlotka S, Rzoska S J, Petrusha I, Stratiichuk D, Turkevich V, Grzegory I 2019 J. Cryst. Growth 505 5
Google Scholar
[39] 孙小伟, 褚衍东, 刘子江, 刘玉孝, 王成伟, 刘维民 2005 物理学报 54 5830
Google Scholar
Sun X W, Chu Y D, Liu Z J, Liu Y X, Wang C W, Liu W M 2005 Acta Phys. Sin. 54 5830
Google Scholar
[40] Saitta A M, Decremps F 2004 Phys. Rev. B 70 035214
Google Scholar
[41] Perlin P, Jauberthie-Carillon C, Itie J P, San Miguel A, Grzegory I I, Polian A 1992 Phys. Rev. B 45 83
Google Scholar
[42] Christensen N E, Gorczyca I I 1994 Phys. Rev. B 50 4397
[43] Muñoz A, Kunc K 1994 Comp. Mater. Sci. 2 400
Google Scholar
-

图 1 GaN 电子局域函数图 (a) 纤锌矿结构(110)晶面; (b) 岩盐结构(100)晶面
Fig. 1. Electron localization function of GaN (a) wurtzite structure (110) and (b) rocksalt structure (100) crystal planes.

图 2 GaN 纤锌矿和岩盐结构在 300 K 下的体积比率与已有结果对比, 所有数据均归一化至纤锌矿结构初始体积, 红色与黑色圆点为分子动力学模拟结果(实线为拟合曲线); 蓝色正三角形与绿色菱形分别为 Xia 等[8]与 Ueno 等[10]的X射线衍射实验结果; 洋红色虚线为 Pandey 等[28]的从头算结果
Fig. 2. The volume ratios of GaN with wurtzite and rocksalt structures at 300 K are compared with existing results, all data are normalized to the initial volume of the wurtzite GaN, the red and black dots are the molecular dynamics simulations results (solid line is the fitted curve). The green diamond and blue square triangle are the X-ray diffraction experimental results by Ueno et al.[10] and Xia et al.[8], respectively. The magenta dashed line is the ab initio result by Pandey et al.[28].
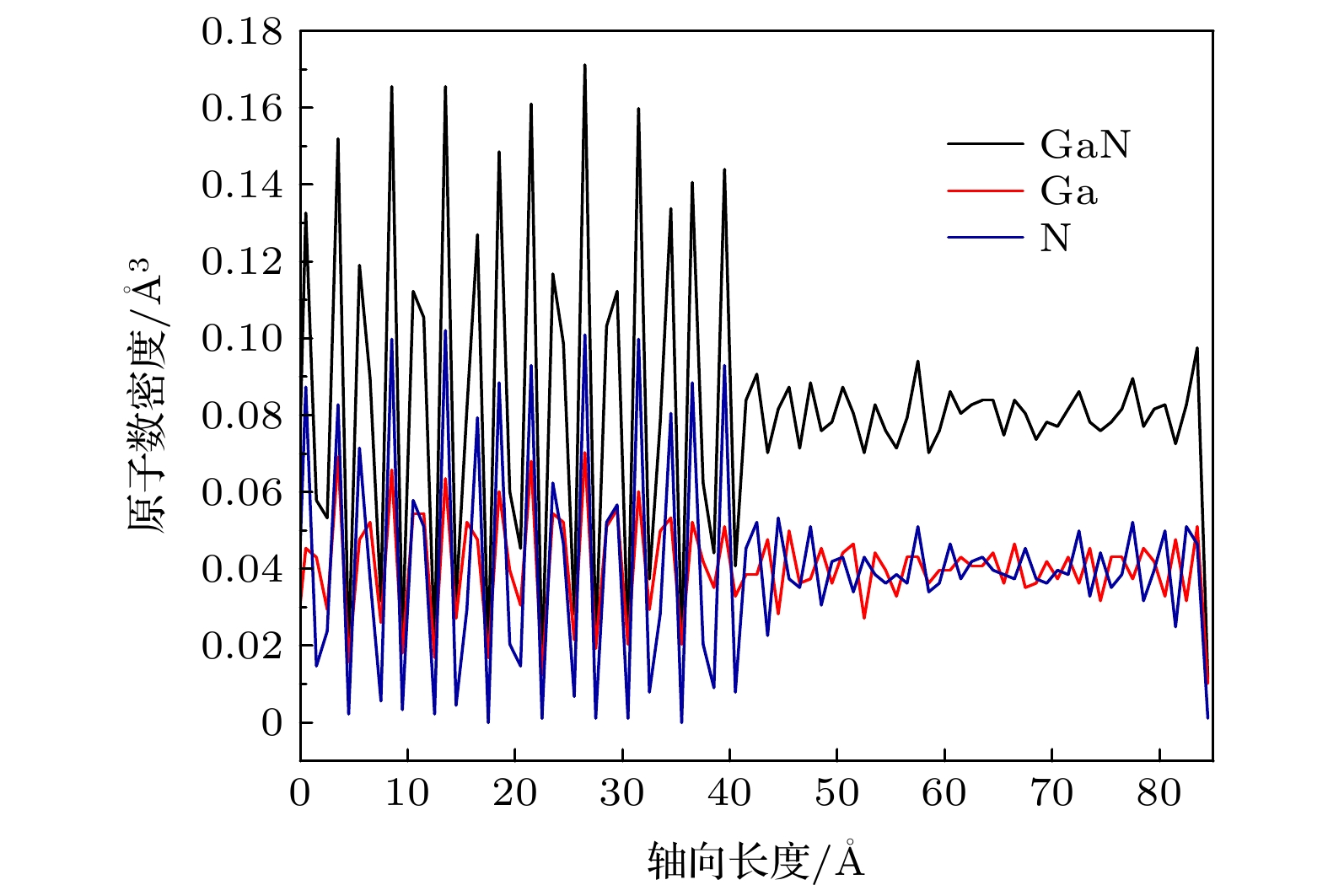
图 4 两相共存状态下 GaN 体系沿 z 轴方向的原子数密度
Fig. 4. Atomic number density along the z-direction of the GaN system in the two-phase coexistence state.
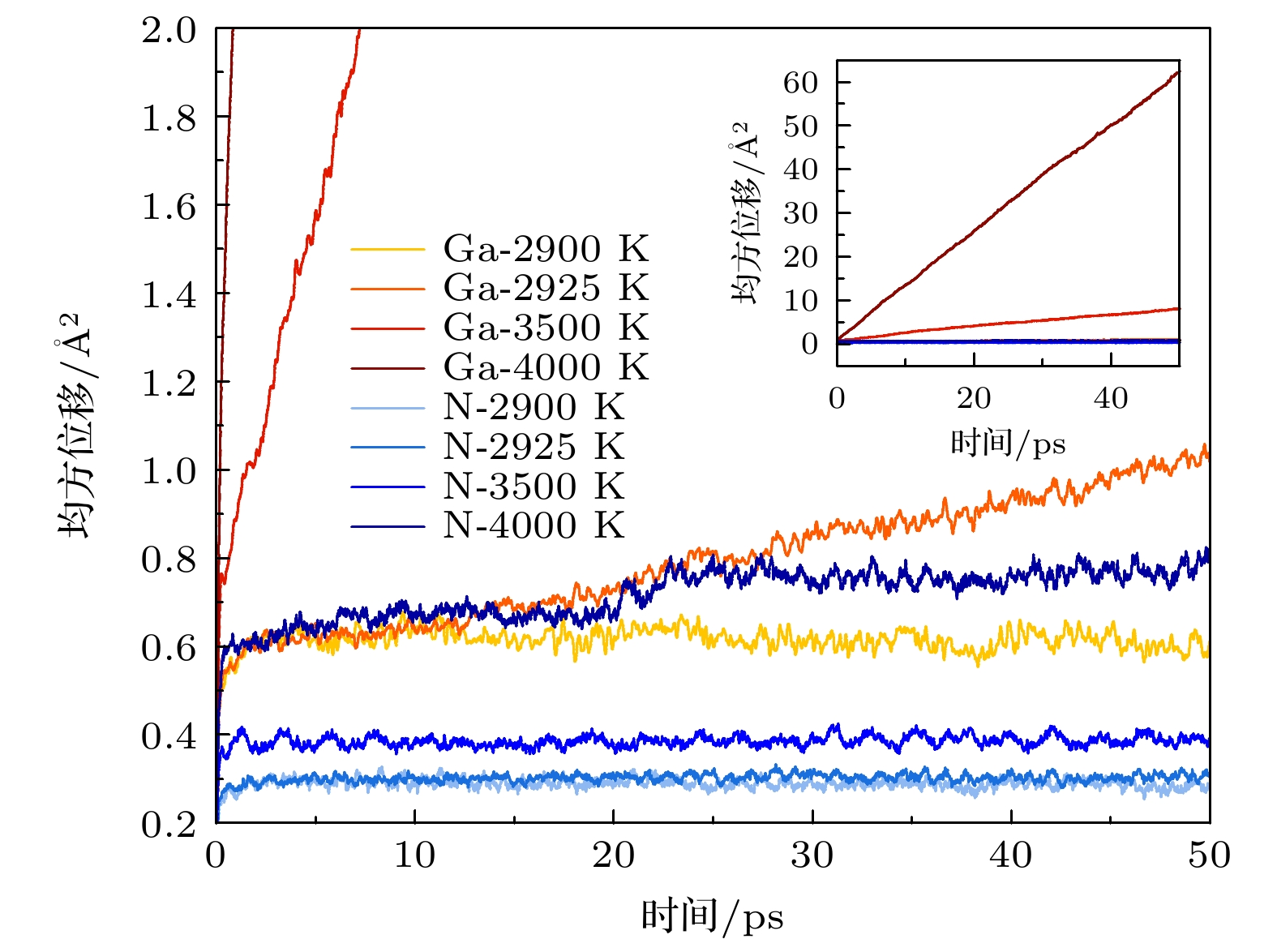
图 5 压力为 10 GPa 时, GaN 在不同温度下的 Ga 原子与 N 原子的均方位移, 内插图为总览图
Fig. 5. Mean square displacement of the Ga and N atoms with different temperatures for GaN at 10 GPa, in which the inset is a general view.

图 6 压力为 10 GPa, 温度为 4000 K 时的 GaN 原子构型, 体系已经处于超离子相 (a) 原始构型; (b) Ga 原子亚晶格; (c) N 原子亚晶格
Fig. 6. GaN in superionic state at 10 GPa and 4000 K (a) original configuration, (b) Ga and (c) N atomic sublattices.
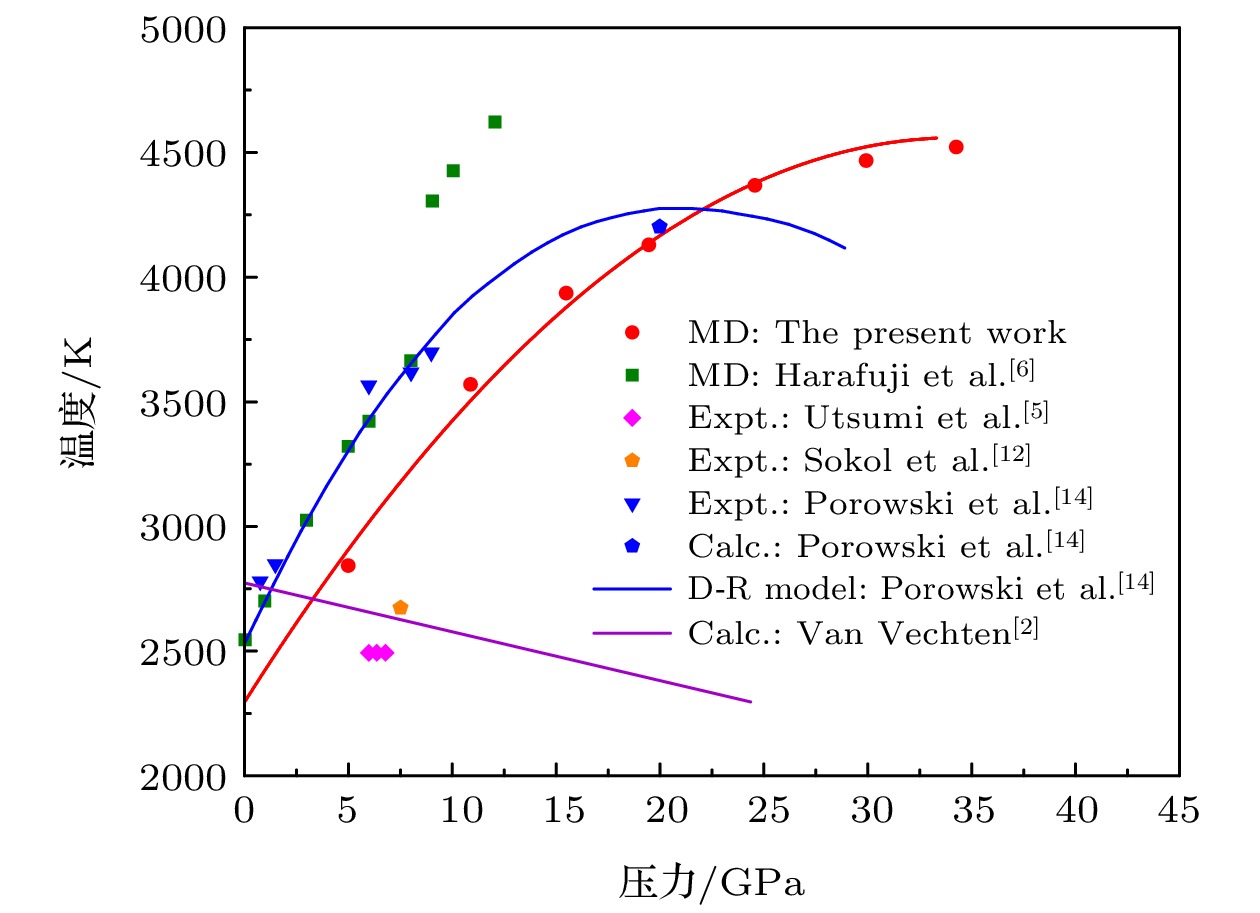
图 7 GaN纤锌矿结构熔化温度的已有研究结果与本文计算结果的对比
Fig. 7. Comparison between existing results on the melting temperature of GaN wurtzite structures and the results calculated in this paper.

图 8 第一性原理计算得到的 GaN 纤锌矿、闪锌矿与岩盐三种结构的相对焓, 内插图为放大的相对焓交点
Fig. 8. Relative enthalpies of GaN with wurtzite, zincblende, and rocksalt structures are calculated by first-principles calculations, and the interpolation shows the enlarged relative enthalpies intersection.

图 9 GaN纤锌矿与岩盐结构固-固相界线的已有数据与本文计算结果的对比
Fig. 9. Comparison of the existing solid-solid phase boundary results of GaN with wurtzite and rocksalt structures with the results calculated in this paper.

图 10 GaN 0—80 GPa 压力范围内的 P-T 相图, 图中的所有点均为分子动力学模拟值, 黑色和红色线为 GaN纤锌矿结构、岩盐结构熔化温度的拟合曲线; 海蓝色与紫色线为 GaN纤锌矿超离子相、岩盐超离子相的转变边界拟合曲线; 蓝色、绿色与橙色点线分别为 GaN纤锌矿超离子相-岩盐超离子相、纤锌矿超离子相-固态岩盐结构、固态纤锌矿结构-固态岩盐结构的相转变边界
Fig. 10. P-T phase diagram of GaN in the pressure range of 0–80 GPa, WZ and RS are used to denote the wurtzite and rocksalt structures of GaN in the diagram. All points are molecular dynamics simulations values in the diagram, the black and red lines are the fitted curves of melting temperature of the GaN with wurtzite and rocksalt structures. The navy blue and purple lines are the fitted curves of phase transition boundary of wurtzite superionic and rocksalt superionic for GaN. The blue, green and orange dotted lines are the phase transition boundary of GaN with wurtzite phase superionic-rocksalt phase superionic, wurtzite phase superionic-rocksalt solid and wurtzite solid-rocksalt solid, respectively.
表 1 GaN的 Morse 势参数与本文拟合得到的 Born-Mayer 势参数
Table 1. The Morse potential parameters and fitted Born-Mayer potential parameters of GaN.
原子对 势函数类型 D/eV γ R/Å A/eV η/Å Z=|Zi|=|Zj| rcut/Å Ga-N Morse[21] 4.5517 6.4764 1.6931 0.63 e 10 Ga-Ga Born-Mayer 227.0715 0.4614 N-N Born-Mayer 531.5281 0.5157  下载: 导出CSV
下载: 导出CSV
表 2 GaN纤锌矿与岩盐结构在零压下的晶格常数, 弹性常数 Cij, 体积模量 B 及晶格能 E
Table 2. The lattice constants, elastic constants Cij, bulk modulus B and lattice energy E of GaN with wurtzite and rocksalt structures at zero pressure.
纤锌矿结构 岩盐结构 DFT LD Ref. DFT LD Ref. a/Å 3.225 3.204 3.188[29] 4.278 4.233 4.278[3] c/Å 5.253 5.102 5.183[29] — — — C11/GPa 326.8 400.3 360.7[30] 323.4 292.8 — C12/GPa 114.7 164.6 115.4[30] 203.7 267.9 — C44/GPa 88.9 118.9 100.1[30] 159.4 267.9 — C13/GPa 82.1 136.4 103.7[30] — — — C33/GPa 362.4 384.4 393.8[30] — — — B/ GPa 174.9 228.6 210.0[31] 214.1 276.2 248.0[8] E/(eV/f.u.) –2.18 –4.35 –2.17[3] –1.23 –3.41 –1.36[3]
下载: 导出CSV
表 3 GaN P-T 相图中三相共存点的位置
Table 3. Location of three-phase coexistence points in GaN P-T phase diagram.
三相共存点 压力/GPa 温度/K P1 33.3 4557 P2 2.0 2550 P3 33.1 4182 P4 36.2 3800
下载: 导出CSV
-
[1] Abdul Amir H A A, Fakhri M A, Abdulkhaleq Alwahib A 2021 Mater. Today Proc. 42 2815
Google Scholar
[2] Van Vechten J A 1973 Phys. Rev. B 7 1479
Google Scholar
[3] Zhou Y, Wang S F, Wang R, Jiang N 2013 Physica B 431 115
Google Scholar
[4] Sadovyi B, Wierzbowska M, Stelmakh S, Boccato S, Gierlotka S, Irifune T, Porowski S, Grzegory I 2020 Phys. Rev. B 102 235109
Google Scholar
[5] Utsumi W, Saitoh H, Kaneko H, Watanuki T, Aoki K, Shimomura O 2003 Nat. Mater. 2 735
Google Scholar
[6] Harafuji K, Tsuchiya T, Kawamura K 2004 J. Appl. Phys. 96 2501
Google Scholar
[7] Karpiński J, Jun J, Porowski S 1984 J. Cryst. Growth 66 1
Google Scholar
[8] Xia H, Xia Q, Ruoff A L 1993 Phys. Rev. B 47 12925
Google Scholar
[9] Novikov S V, Zainal N, Akimov A V, Staddon C R, Kent A J, Foxon C T 2010 J. Vac. Sci. Technol. B 28 C3B1
Google Scholar
[10] Ueno M, Yoshida M, Onodera A, Shimomura O, Takemura K 1994 Phys. Rev. B 49 14
Google Scholar
[11] Saoud F S, Plenet J C, Louail L, Maouche D 2011 Comput. Theor. Chem. 964 65
Google Scholar
[12] Sokol A G, Palyanov Y N, Surovtsev N V 2007 Diam. Relat. Mater. 16 431
Google Scholar
[13] Saitoh H, Utsumi W, Kaneko H, Aoki K 2007 J. Cryst. Growth 300 26
Google Scholar
[14] Porowski S, Sadovyi B, Gierlotka S, Rzoska S J, Grzegory I, Petrusha I, Turkevich V, Stratiichuk D 2015 J. Phys. Chem. Solids 85 138
Google Scholar
[15] Nord J, Albe K, Erhart P, Nordlund K 2003 J. Phys. Condens. Matter 15 5649
Google Scholar
[16] Alder B J, Wainwright T E 1957 J. Chem. Phys. 27 1208
Google Scholar
[17] Kioseoglou J, Polatoglou H M, Lymperakis L, Nouet G, Komninou P 2003 Comp. Mater. Sci. 27 43
Google Scholar
[18] Béré A, Serra A 2006 Philos. Mag. 86 2159
Google Scholar
[19] Do E C, Shin Y H, Lee B J 2009 J. Phys. :Condens. Matter 21 325801
Google Scholar
[20] Zapol P, Pandey R, Gale J D 1997 J. Phys. :Condens. Matter 9 9517
Google Scholar
[21] Zhang S, Chen N X 2005 Chem. Phys. 309 309
Google Scholar
[22] Born M, Mayer J E 1932 Z. Physik 75 1
Google Scholar
[23] Fischer T H, Almlof J 1992 J. Phys. Chem. 96 9768
Google Scholar
[24] Perdew J P, Ruzsinszky A, Csonka G I, Vydrov O A, Scuseria G E, Constantin L A, Zhou X, Burke K 2008 Phys. Rev. Lett. 100 136406
Google Scholar
[25] Vanderbilt D 1990 Phys. Rev. B 41 7892
Google Scholar
[26] Clark S J, Segall M D, Pickard C J, Hasnip P J, Probert M I, Refson K, Payne M C 2005 Z. Kristallogr. 220 567
[27] Gale J D, Rohl A L 2003 Mol. Simulat. 29 291
Google Scholar
[28] Pandey R, Jaffe J E, Harrison N M 1994 J. Phys. Chem. Solids 55 1357
Google Scholar
[29] Kirchner V, Heinke H, Hommel D, Domagala J Z, Leszczynski M 2000 Appl. Phys. Lett. 77 1434
Google Scholar
[30] Adachi K, Ogi H, Nagakubo A, Nakamura N, Hirao M, Imade M, Yoshimura M, Mori Y 2016 J. Appl. Phys. 119 245111
Google Scholar
[31] Polian A, Grimsditch M, Grzegory I 1996 J. Appl. Phys. 79 3343
Google Scholar
[32] Plimpton S 1995 J. Comput. Phys. 117 1
Google Scholar
[33] Zou Y C, Xiang S K, Dai C D 2020 Comp. Mater. Sci. 171 109156
Google Scholar
[34] Hazarika M P, Chakraborty S N 2019 Chem. Phys. Lett. 730 521
Google Scholar
[35] Liu C M, Xu C, Cheng Y, Chen X R, Cai L C 2015 J. Appl. Phys. 118 235901
Google Scholar
[36] Wang S C, Zhang G M, Liu H F, Song H F 2013 J. Chem. Phys. 138 134101
Google Scholar
[37] Cazorla C, Errandonea D 2013 J. Phys. Chem. C 117 11292
[38] Porowski S, Sadovyi B, Karbovnyk I, Gierlotka S, Rzoska S J, Petrusha I, Stratiichuk D, Turkevich V, Grzegory I 2019 J. Cryst. Growth 505 5
Google Scholar
[39] 孙小伟, 褚衍东, 刘子江, 刘玉孝, 王成伟, 刘维民 2005 物理学报 54 5830
Google Scholar
Sun X W, Chu Y D, Liu Z J, Liu Y X, Wang C W, Liu W M 2005 Acta Phys. Sin. 54 5830
Google Scholar
[40] Saitta A M, Decremps F 2004 Phys. Rev. B 70 035214
Google Scholar
[41] Perlin P, Jauberthie-Carillon C, Itie J P, San Miguel A, Grzegory I I, Polian A 1992 Phys. Rev. B 45 83
Google Scholar
[42] Christensen N E, Gorczyca I I 1994 Phys. Rev. B 50 4397
[43] Muñoz A, Kunc K 1994 Comp. Mater. Sci. 2 400
Google Scholar

 下载:
下载:

计量
- 文章访问数: 10001
- PDF下载量: 185
- 被引次数: 0
