










-
经式8-羟基喹啉铝(mer-Alq3)是一种光电性能优良的小分子有机半导体发光材料. 本文采用密度泛函理论(DFT)B3LYP/6-31G*方法和基组对其进行结构优化, 计算并研究了该分子的红外光谱、拉曼光谱和前线轨道. 计算得到的红外光谱、拉曼光谱均与实验相符. 前线轨道表明基态最高占据轨道(HOMO)的电子云主要集中在苯酚环, 最低未占据轨道(LUMO)的电子云主要集中在吡啶环. 用含时密度泛函理论(TD-DFT)计算得到紫外-可见吸收光谱, 采用空穴-电子分析法研究了电子激发特征. 结果表明: 电子从基态到激发态的跃迁, 主要是8-羟基喹啉环内或环间的电荷转移, 以π-π*跃迁为主, 包括局域激发和电荷转移激发两种类型. 本工作对mer-Alq3分子发光机理提出更深入的认识, 能为进一步提高该分子发光效率和调控分子的发光范围提供一定的理论指导.Meridional tris(8-hydroxyquinoline)aluminum (III) (mer-Alq3) is an organometallic semiconductor material with phenomenal photo-electric properties. In order to understand the molecular luminescence properties of mer-Alq3, the density functional theoretical (B3LYP) method with 6-31G* basis set is employed to calculate its structure, infrared spectrum and Raman spectrum and the frontier molecular orbital of its ground state. The UV-vis absorption and the excited state characteristics are investigated by the time-dependent density functional theory (TD-DFT) method. The results show that the calculated spectral characteristics are in good agreement with the experimental data. The electron cloud of the highest occupied molecular orbital (HOMO) is located mostly on the phenoxide ring, whereas that of the lowest unoccupied molecular orbital (LUMO) sits on the pyridine ring. The absorption peaks of the UV-visible absorption spectrum are located in the visible and ultraviolet region. S0→S2 is attributed to the superposition of the π-π* local excitation in the direction from benzene ring to pyridine ring and the n-π* local excitation in the direction from oxygen atom to pyridine ring. The π-π* local excitation from benzene ring to pyridine ring is S0→S4. The superposition of π-n local excitation from benzene to carbon and n-n local excitation from oxygen to carbon are excited by S0→S11. S0→S14 is charge-transfer excitation and contributed by the superposition of π-π* in the direction from benzene ring to pyridine ring and n-π* in the direction from oxygen atom to pyridine ring. This work is significant for understanding the basic properties of mer-Alq3 and the mechanisms of electron excitations. It provides a deeper insight into the luminescence mechanism of mer-Alq3, thus playing a guidance role in further improving the luminescence efficiency and regulating the spectral range of the light-emitting mer-Alq3.
-
Keywords:
- density functional theory /
- mer-Alq3 /
- spectral properties /
- excited state
[1] 段炼, 邱勇 2015 材料研究学报 29 321
 Google Scholar
Google Scholar
Duan L, Qiu Y 2015 Chin. J. Mater. Res. 29 321
Google Scholar
[2] 周瑞, 安忠维, 柴生勇 2004 光谱学与光谱分析 08 922
Google Scholar
Zhou R, An Z W, Chai S Y 2004 Spectrosc. Spect. Anal. 08 922
Google Scholar
[3] Xu H, Chen R F, Sun Q, Lai W Y, Su Q Q, Huang W, Liu X G 2014 Chem. Soc. Rev. 43 3259
Google Scholar
[4] Tang C W, VanSlyke S A 1987 Appl. Phys. Lett. 51 913
Google Scholar
[5] Liao S H, Shiu J R, Liu S W, Yeh S J, Chen Y H, Chen C T, Chow T J, Wu C I 2009 J. Am. Chem. Soc. 131 763
Google Scholar
[6] Liu R, Gan Z Q, Shinar R, Shinar J 2011 Phys. Rev. B 83 245302
Google Scholar
[7] Katakura R, Koide Y 2006 Inorg. Chem. 45 5730
Google Scholar
[8] 许金钩, 王尊本 2006 荧光分析法 (北京: 科学出版社) 第42页
Xu J G, Wang Z B 2006 Fluorescence Analysis (Beijing: Science Press) p42 (in Chinese)
[9] 张蕾, 张学俊 2011 化工中间体 7 28
Zhang L, Zhang X J 2011 Chem. Intermed. 7 28
[10] Xu B S, Chen L Q, Liu X G, Zhou H F, Xu H F, Fang X H, Wang Y L 2008 Appl. Phys. Lett. 92 103305
Google Scholar
[11] Pérez-Bolívar C, Takizawa S Y, Nishimura G, Montes V A, Anzenbacher P J 2011 Chem. Eur. J. 17 9076
Google Scholar
[12] Stampor W, Kalinowski J, Marco P D, Fattori V 1997 Appl. Phys. Lett. 70 1953
Google Scholar
[13] Xue W M, Wang Y, Chan M C W, Su Z M, Cheung K K, Chen C M 1998 Organomet. 17 1946
Google Scholar
[14] Xue W M, Chan M C W, Su Z M, Cheung K K, Liu S T, Chen C M 1998 Organomet. 17 1622
Google Scholar
[15] 苏忠民, 高洪泽, 程红, 初蓓, 陈丽华, 王荣顺, 王悦, 沈家骢 2001 中国科学: 化学 31 16
Su Z M, Gao H Z, Chen H, Chu B, Chen L H, Wang R S, Wang Y, Shen J C 2001 Sci. China: Chemistry 31 16
[16] Curioni A, Boero M, Andreoni W 1998 Chem. Phys. Lett. 294 263
Google Scholar
[17] Sobereva http://sobereva.com/434 [2019-10-25]
[18] Tangui L B, Carlo A, Ilaria C 2011 J. Chem. Theory Comput. 7 2498
Google Scholar
[19] Foresman J B, Frisch A 1996 Exploring chemistry with electronic structure method (2nd edn.) (Pittsburgh: Gaussian, Inc.) p64
[20] Curioni A, Andreoni W 2001 IBM J. Res. Dev. 45 101
Google Scholar
[21] Brinkmann M, Gadret G, Muccini M, Taliani C, Masciocchi N, Sironi A 2000 J. Am. Chem. Soc. 122 5147
Google Scholar
[22] Chemistry Database [DB/OL]. Shanghai Institute of Organic Chemistry of CAS. http://www.organchem.csdb.cn. [1978–2019]
[23] 王媛媛 2006 硕士学位论文 (山东: 山东大学)
Wang Y Y 2006 M.S. Thesis (Shangdong: Shandong University) (in Chinese)
[24] 解晓东, 郝玉英, 章日光, 王宝俊 2012 物理学报 61 127201
Google Scholar
Xie X D, Hao Y Y, Zhang R G, Wang B J 2012 Acta Phys. Sin. 61 127201
Google Scholar
[25] 卢天, 陈飞武 2011 化学学报 69 2393
Lu T, Chen F W 2011 Acta Chim. Sin. 69 2393
[26] Lu T, Chen F W 2012 J. Comput. Chem. 33 580
Google Scholar
[27] Multiwfn Manual, Lu T http://sobereva.com/multiwfn/[2019-9-2]
-
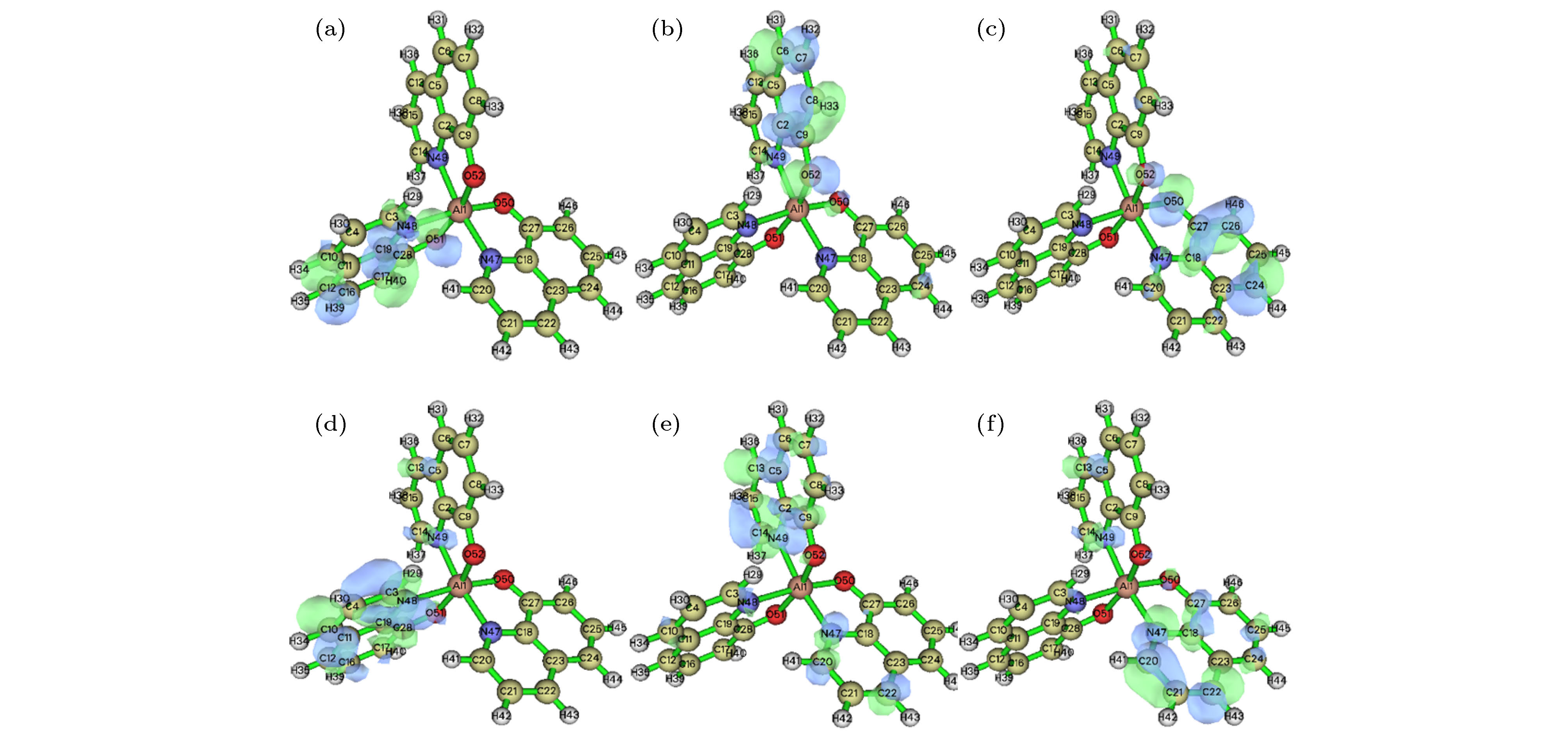
图 4 mer-Alq3前线分子轨道分布图 (a) HOMO-2轨道分布图; (b) HOMO-1轨道分布图; (c) HOMO轨道分布图; (d) LUMO轨道分布图; (e) LUMO+1轨道分布图; (f) LUMO+2轨道分布图
Fig. 4. Frontier molecular orbits of mer-Alq3: (a) HOMO-2 distribution; (b) HOMO-1 distribution; (c) HOMO distribution; (d) LUMO distribution; (e) LUMO+1 distribution; (f) LUMO+2 distribution.

图 6 mer-Alq3的空穴-电子、Chole-Cele、Sr示意图 (a)−(c) S2的空穴-电子, Chole-Cele, Sr图; (d)−(f) S4的空穴-电子, Chole-Cele, Sr图; (g)−(i) S11的空穴-电子, Chole-Cele, Sr图; (j)−(l) S14的空穴-电子, Chole-Cele, Sr图
Fig. 6. Electron-hole, Chole-Cele and Sr
distributions of mer-Alq3 respectively: (a)−(c) Electron-hole, Chole-Cele, Sr distribution at S2 state geometry; (d)−(f) Electron-hole, Chole-Cele, Sr distribution at S4 state geometry; (g)−(i) Electron-hole, Chole-Cele, Sr distribution at S11 state geometry; (j)−(l) Electron-hole, Chole-Cele, Sr distribution at S14 state geometry 表 1 mer-Alq3分子的键长
Table 1. Bond lengths of the mer-Alq3.
Bond B3LYP/6-31G*/Å Experimental results/Å[21] Al-Na 2.08377 2.0502 Al-Nb 2.12565 2.0872 Al-Nc 2.06431 2.0172 Al-Oa 1.85545 1.8502 Al-Ob 1.88106 1.8602 Al-Oc 1.88398 1.8572  下载: 导出CSV
下载: 导出CSV
表 2 mer-Alq3分子中部分振动模式指认
Table 2. Identification of partial vibration modes of mer-Alq3.
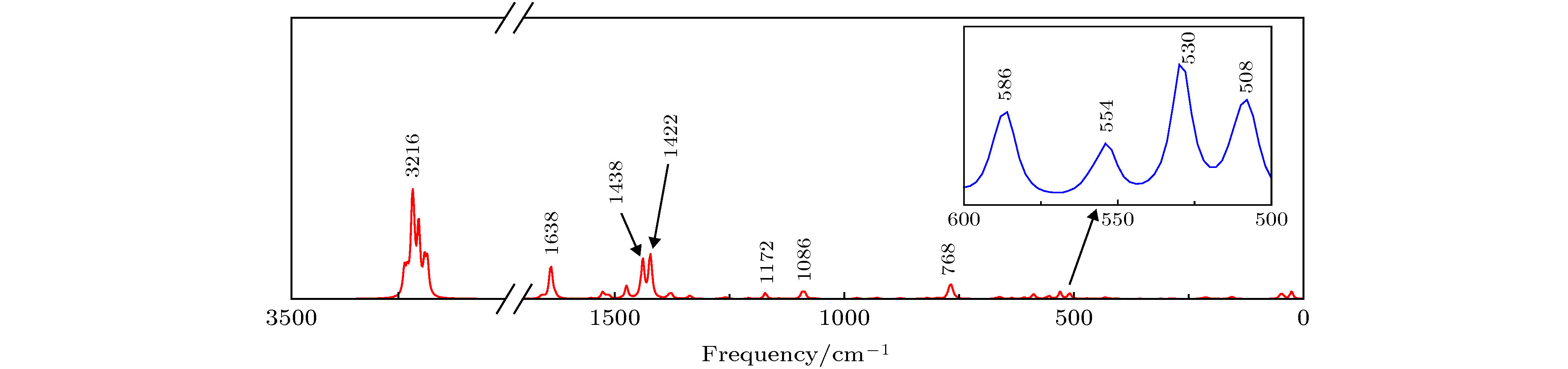
Vexperiment/cm–1 Vtheory/cm–1 Vibration analysis Vexperiment/cm–1 Vtheory/cm–1 Vibration analysis 398 408 分子骨架扭曲变形 1228 1268 C—N伸缩振动, C—H平面摇摆振动, 剪式振动 416 422 分子骨架扭曲变形, 环1环2环5 环6上C—H平面摇摆振动, 环3环4上C—H扭曲振动 1280 1334 C—O, C—C伸缩振动, C—H平面摇摆振动 457 470 C—H扭曲振动 1328 1376 C—H平面摇摆振动, 剪式振动 548 554 Al-O50伸缩振动, 环1环2呼吸振动 1383 1422 C—C伸缩振动, C—H平面摇摆振动 642 662 Al-O50伸缩振动, C—H扭曲振动 1424 1438 C—N、C—C伸缩振动, C—H 平面摇摆振动, 剪式振动 746 768 Al-O面外弯曲振动, 环3环4呼吸
振动1468 1512 C—N伸缩振动, C—C伸缩振动, C—H平面摇摆振动 787 796 C—H扭曲振动 1499 1550 C—C伸缩振动, C—H平面摇摆振动 803 820 苯环和吡啶环变形振动 1579 1636 C—N伸缩振动, C—C伸缩振动, C—H平面摇摆振动, 剪式振动 823 836 C—H面外摇摆振动 1606 1658 C—N伸缩振动, C—C伸缩振动, C—H平面摇摆振动, 剪式振动 1114 1140 C—H剪式振动 3039 3202 苯环上C—H伸缩振动
下载: 导出CSV
表 3 mer-Alq3分子中部分振动模式指认
Table 3. Identification of partial vibration modes of mer-Alq3.
Vexperiment/cm–1 Vtheory/cm–1 Vibration analysis 507 508 Al—O扭曲振动, 苯环和吡啶环变形振动 529 530 Al—O伸缩振动, 苯环和吡啶环呼吸振动 545 554 Al—O伸缩振动, 苯环和吡啶环呼吸振动 581 586 Al—O扭曲振动, 苯环和吡啶环变形振动 760 768 Al—O伸缩振动, 苯环和吡啶环呼吸振动 1062 1086 C—H平面摇摆振动, 剪式振动 1177 1172 C—H平面摇摆振动, 剪式振动 1393 1422 C—O, C—C伸缩振动, C—H平面摇摆振动, 剪式振动 — 1438 C—N、C—C伸缩振动, C—H平面摇摆振动, 剪式振动 1593 1638 C—C伸缩振动, C—H平面摇摆振动, 剪式振动 — 3216 C—H伸缩振动
下载: 导出CSV
表 4 mer-Alq3的前线分子轨道能级(单位: arb.units)及分布(单位: %)
Table 4. Frontier molecular orbital energy levels (in arb.units) and distribution (in %) of mer-Alq3.
分子轨道 能级 Al a b c O 苯 吡啶 O 苯 吡啶 O 苯 吡啶 H-2 –0.19584 1.51 0.16 0.85 0.93 20.36 64.59 19.94 0.24 0.74 1.15 H-1 –0.19204 1.57 2.05 8.50 2.56 0.36 1.18 1.17 17.19 58.30 17.72 H –0.18397 1.47 19.25 57.52 17.84 0.36 0.20 0.24 3.58 7.26 2.42 L –0.06363 1.60 0.06 0.18 0.91 1.66 25.28 64.81 0.29 4.55 12.18 L+1 –0.05496 1.36 0.62 8.41 20.90 0.24 3.24 7.25 1.33 19.64 47.98 L+2 –0.05218 1.28 1.72 21.57 56.03 0.54 1.83 5.13 0.81 6.32 16.59
下载: 导出CSV
表 5 mer-Alq3分子的电子激发分析表
Table 5. The analysis of electron excitation of mer-Alq3.
Excited state λ/nm f Transition nature (contribution > 10%) Transition energy/eV 2 427.15 0.0672 119→121 (45.9956%); 119→122 (23.0683%);
118→120 (21.1263%)2.9026 4 417.31 0.0425 117→120 (88.1022%) 2.9710 11 304.03 0.0151 119→124 (38.2445%); 119→125 (23.0208%) 4.0781 12 302.87 0.0214 117→123 (66.2078%); 114→120 (20.1638%) 4.0937
下载: 导出CSV
表 6 mer-Alq3分子的激发态参数
Table 6. Excited state parameters of mer-Alq3.
D/Å Sr/arb.units H/Å t/Å S0 → S2 0.18 0.61 3.57 0.12 S0 → S4 0.99 0.59 2.95 0.41 S0 → S11 0.88 0.79 3.84 –1.38 S0 → S14 0.68 0.43 3.47 2.00
下载: 导出CSV
-
[1] 段炼, 邱勇 2015 材料研究学报 29 321
Google Scholar
Duan L, Qiu Y 2015 Chin. J. Mater. Res. 29 321
Google Scholar
[2] 周瑞, 安忠维, 柴生勇 2004 光谱学与光谱分析 08 922
Google Scholar
Zhou R, An Z W, Chai S Y 2004 Spectrosc. Spect. Anal. 08 922
Google Scholar
[3] Xu H, Chen R F, Sun Q, Lai W Y, Su Q Q, Huang W, Liu X G 2014 Chem. Soc. Rev. 43 3259
Google Scholar
[4] Tang C W, VanSlyke S A 1987 Appl. Phys. Lett. 51 913
Google Scholar
[5] Liao S H, Shiu J R, Liu S W, Yeh S J, Chen Y H, Chen C T, Chow T J, Wu C I 2009 J. Am. Chem. Soc. 131 763
Google Scholar
[6] Liu R, Gan Z Q, Shinar R, Shinar J 2011 Phys. Rev. B 83 245302
Google Scholar
[7] Katakura R, Koide Y 2006 Inorg. Chem. 45 5730
Google Scholar
[8] 许金钩, 王尊本 2006 荧光分析法 (北京: 科学出版社) 第42页
Xu J G, Wang Z B 2006 Fluorescence Analysis (Beijing: Science Press) p42 (in Chinese)
[9] 张蕾, 张学俊 2011 化工中间体 7 28
Zhang L, Zhang X J 2011 Chem. Intermed. 7 28
[10] Xu B S, Chen L Q, Liu X G, Zhou H F, Xu H F, Fang X H, Wang Y L 2008 Appl. Phys. Lett. 92 103305
Google Scholar
[11] Pérez-Bolívar C, Takizawa S Y, Nishimura G, Montes V A, Anzenbacher P J 2011 Chem. Eur. J. 17 9076
Google Scholar
[12] Stampor W, Kalinowski J, Marco P D, Fattori V 1997 Appl. Phys. Lett. 70 1953
Google Scholar
[13] Xue W M, Wang Y, Chan M C W, Su Z M, Cheung K K, Chen C M 1998 Organomet. 17 1946
Google Scholar
[14] Xue W M, Chan M C W, Su Z M, Cheung K K, Liu S T, Chen C M 1998 Organomet. 17 1622
Google Scholar
[15] 苏忠民, 高洪泽, 程红, 初蓓, 陈丽华, 王荣顺, 王悦, 沈家骢 2001 中国科学: 化学 31 16
Su Z M, Gao H Z, Chen H, Chu B, Chen L H, Wang R S, Wang Y, Shen J C 2001 Sci. China: Chemistry 31 16
[16] Curioni A, Boero M, Andreoni W 1998 Chem. Phys. Lett. 294 263
Google Scholar
[17] Sobereva http://sobereva.com/434 [2019-10-25]
[18] Tangui L B, Carlo A, Ilaria C 2011 J. Chem. Theory Comput. 7 2498
Google Scholar
[19] Foresman J B, Frisch A 1996 Exploring chemistry with electronic structure method (2nd edn.) (Pittsburgh: Gaussian, Inc.) p64
[20] Curioni A, Andreoni W 2001 IBM J. Res. Dev. 45 101
Google Scholar
[21] Brinkmann M, Gadret G, Muccini M, Taliani C, Masciocchi N, Sironi A 2000 J. Am. Chem. Soc. 122 5147
Google Scholar
[22] Chemistry Database [DB/OL]. Shanghai Institute of Organic Chemistry of CAS. http://www.organchem.csdb.cn. [1978–2019]
[23] 王媛媛 2006 硕士学位论文 (山东: 山东大学)
Wang Y Y 2006 M.S. Thesis (Shangdong: Shandong University) (in Chinese)
[24] 解晓东, 郝玉英, 章日光, 王宝俊 2012 物理学报 61 127201
Google Scholar
Xie X D, Hao Y Y, Zhang R G, Wang B J 2012 Acta Phys. Sin. 61 127201
Google Scholar
[25] 卢天, 陈飞武 2011 化学学报 69 2393
Lu T, Chen F W 2011 Acta Chim. Sin. 69 2393
[26] Lu T, Chen F W 2012 J. Comput. Chem. 33 580
Google Scholar
[27] Multiwfn Manual, Lu T http://sobereva.com/multiwfn/[2019-9-2]


 下载:
下载:


计量
- 文章访问数: 14580
- PDF下载量: 154
- 被引次数: 0
