










-
光谱选择性吸收涂层是太阳能光-热利用技术的核心部件, 直接决定着整个系统的转换效率, 为了提高涂层的选择吸收性和热稳定性, 本文提出以金属氮化物替代金属纳米颗粒, 构建纳米晶-非晶异质结构的思路, 并采用多弧离子镀制备了Cr/CrAlN/CrAlON/CrAlN/CrAlON/CrAlO多吸收层光谱选择性吸收涂层, 其吸收率达0.90, 发射率为0.15, 而且在500 ℃、大气条件下时效220 h后, 涂层的吸收率升至0.94, 发射率则降至0.10, 并且能够保持稳定1000 h以上. 微观组织分析表明, 高温时效处理后, 吸收层发生部分晶化形成了大量氮化物纳米颗粒, 增加了对太阳光的散射和吸收, 而CrAlO减反射层中的部分晶化形成了Al2O3和Cr2O3纳米颗粒, 这不仅可以保护内部涂层不被氧化, 而且Al2O3的形成可以增加太阳光的透过率, 减少涂层表面反射, 是多吸收层CrAlON基光谱选择性吸收涂层选择吸收性能提高的主要原因. 同时, 氮化物纳米颗粒被非晶基体均匀地分隔开来, 形成了纳米晶-非晶异质结构, 非晶在高温时效处理过程中只发生结构弛豫, 从而有效地抑制了高温条件下的原子扩散, 保证涂层中的纳米颗粒在高温下不发生明显团聚, 这是多吸收层CrAlON基涂层具有良好热稳定性的最主要原因. 这些研究结果对提高金属陶瓷光谱选择性吸收涂层的综合性能, 实现更高效率的太阳能光-热利用具有重大意义.Spectrally selective absorbing coating is the core component of the utilization of solar energy. The spectral properties of selectively absorbing coating directly determine the conversion efficiency of constructing solar power plants. To enhance the selective absorbability and thermal stability, we propose an idea that these metal particles are replaced with transition-metal nitrides, and then coated with periodic nanocrystalline-amorphous heterogeneous structures. Double-absorbing layer Cr/CrAlN/CrAlON/CrAlN/CrAlON/CrAlO solar selective absorbing coatings with a high solar absorptance of 0.90 and a relatively low emittance of 0.15 are obtained by the cathodic arc ion plating technique. After the coating is aged at 500 °C in air for 220 h, its absorptance increases to 0.94 and the emittance decreases to 0.10. More importantly, the coating exhibits an outstanding thermal stability with a selectivity of 0.94/0.11 even after being aged at 500 °C for 1000 h in air. The microstructure analysis indicates that the multilayer coating consists of aperiodic CrAlN and CrAlON layers in addition to the Cr and CrAlO layers. Through the long-term aging, a small number of AlN, CrN and Cr2N nanocrystallites are observed to be homogeneously embedded in the CrAlN and CrAlON amorphous matrices. The nanoparticles in the CrAlN and CrAlON layers can effectively scatter the incident light into a broadband wavelength range, increasing the optical path length in the absorbing layers, and thus resulting in a pronounced enhancement in the absorptivity. A handful of Cr2O3 and Al2O3 nanograins are observed to be embedded in the amorphous CrAlO antireflection layer, which can effectively reflect the solar infrared radiation and the thermal emittance from the substrate, and thus resulting in pretty low infrared emissivity. The good thermal stability is attributed to the excellent thermal stability of the dielectric amorphous matrices and the sluggish atomic diffusion in the nanoparticles, which could effectively slow down the inward diffusion of oxygen and avoid agglomerating the nanoparticles. These results are of great importance for enhancing the overall performance of cermet spectrally selective absorption coating and also for improving the conversion efficiency of solar energy photo-thermal utilization.
-
Keywords:
- spectrally selective absorbing coating /
- thermal stability /
- absorptance /
- emittance
[1] 史月艳, 那鸿悦 2009 太阳光谱选择性吸收膜系设计、制备及测评 (第1版) (北京: 清华大学出版社) 第44−65页
Shi Y Y, Na H Y 2009 Design, Preparation and Evaluation of Solar Spectrum Selective absorption Films (1st Ed.) (Beijing: Tsinghua University Press) pp44−65 (in Chinese)
[2] Cao F, McEnaney K, Chen G, Ren Z F 2014 Energy Environ. Sci. 7 1615
 Google Scholar
Google Scholar
[3] Cao F, Kraemer D, Sun T Y, Lan Y C, Chen G, Ren Z F 2015 Adv. Energy Mater. 5 1401042
Google Scholar
[4] Wang X Y, Gao J H, Hu H B, Zhang H L, Liang L Y, Javaid K, Zhuge F, Cao H T, Wang L 2017 Nano Energy 37 232
Google Scholar
[5] Xue Y F, Wang C, Wang W W, Liu Y, Wu Y X, Ning Y P, Sun Y 2013 Sol. Energy 96 113
Google Scholar
[6] Cheng J S, Wang C, Wang W W, Du X K, Liu Y, Xue Y F, Wang T M, Chen B L 2013 Sol. Energy Mater. Sol. Cells 109 204
Google Scholar
[7] 田广科, 苗树翻, 马天国, 范多旺 2015 太阳能 3 50
Google Scholar
Tian G K, Mi ao, Ma T G, Fan D W 2015 Sol. Energy 3 50
Google Scholar
[8] Ge J P, Zhang Q, Zhang T R, Yin Y D 2008 Angew. Chem. Int. Edit. 47 8924
Google Scholar
[9] Joo S H, Park J Y, Tsung C K, Yamada Y, Yang P, Somorjai G A 2008 Nat. Mater. 8 126
Google Scholar
[10] Gao T, Jelle B P, Gustavsen A J 2013 Nanopart. Res. 15 1370
Google Scholar
[11] Cao A, Veser G 2009 Nat. Mater. 9 75
Google Scholar
[12] Kim T K, VanSaders B, Caldwell E, Shin S, Liu Z, Jin S, Chen R 2016 Sol. Energy 132 257
Google Scholar
[13] Liu H D, Wan Q, Xu Y R, luo C, Chen Y M, Fu D J, Ren F, Luo G, Cheng X D, Hu X J, Yang B 2015 Sol. Energy Mater. Sol. Cells 134 261
Google Scholar
[14] Wu L, Gao J H, Liu Z M, Liang L Y, Xia F, Cao H T 2013 Sol. Energy Mater. Sol. Cells 114 186
Google Scholar
[15] Du M, Hao L, Mi J, Lü F, Liu X P, Jiang L J, Wang S M 2011 Sol. Energy Mater. Sol. Cells 95 1193
Google Scholar
[16] Barshilia H C 2014 Sol. Energy Mater. Sol. Cells 130 322
Google Scholar
[17] Barshilia H C, Selvakumar N, Rajam K S, Biswas A 2008 Sol. Energy Mater. Sol. Cells 92 1425
Google Scholar
[18] Barshilia H C, Selvakumar N, Rajam K S 2007 J. Vac. Sci. Technol., A 25 383
Google Scholar
[19] 史月艳, 那鸿悦 2009 太阳光谱选择性吸收膜系设计、制备及测评 (第1版) (北京: 清华大学出版社) 第67−147页
Shi Y Y, Na H Y 2009 Design, Preparation and Evaluation of Solar Spectrum Selective absorption Films (1st Ed.) (Beijing: Tsinghua University Press) pp67−147(in Chinese)
[20] Zou C W, Xie W, Shao L X 2016 Sol. Energy Mater. Sol. Cells 153 9
Google Scholar
[21] Liu H D, Fu T R, Duan M H, Wan Q, luo C, Chen Y M, Fu D J, Ren F, Li Q Y, Cheng X D, Yang B, Hu X J 2016 Sol. Energy Mater. Sol. Cells 157 108
Google Scholar
[22] Gong D Q, Liu H D, Luo G, Zhang P, Cheng X D, Yang B, Wang Y B, Min J, Wang W X, Chen S P, Cui Z Q, Li K W, Hu L F 2015 Sol. Energy Mater. Sol. Cells 136 167
Google Scholar
[23] Gammer C, Mangler C, Rentenberger C, Karnthaler H P 2010 Scr. Mater. 63 312
Google Scholar
[24] Gao X H, Guo Z M, Geng Q F, Ma P J, Wang A Q, Liu G 2016 Sol. Energy 140 199
Google Scholar
[25] van den Oetelaar L C A, Nooij O W, Oerlemans S, Denier van der Gon A W, Brongersma H H, Lefferts L, Roosenbrand A G, van Veen J A R 1998 J. Phys. Chem. B 102 3445
Google Scholar
[26] Malis O, Radu M, Mott D, Wanjala B, Luo J, Zhong C J 2009 Nanotechnology 20 245708
Google Scholar
[27] Liao H, Fisher A, Xu Z J 2015 Small 11 3221
Google Scholar
[28] Dean J A 1990 Mater. Manuf. Processes 5 687
Google Scholar
[29] Clark B G, Hattar K, Marshall M T, Chookajorn T, Boyce B L, Schuh C A 2016 JOM 68 1625
Google Scholar
[30] Han L L, Meng Q P, Wang D L, Zhu Y M, Wang J, Du X W, Stach E A, Xin H L 2016 Nat. Commun. 7 13335
Google Scholar
[31] Huolin L X, Selim A, Runzhe T, Arda G, Chong-Min W, Libor K, Eric A S, Lin-Wang W, Miquel S, Gabor A S, Haimei Z 2014 Nano Lett. 14 3203
Google Scholar
[32] Wang C M, Genc A, Cheng H, Pullan L, Baer D R, Bruemmer S M 2014 Sci. Rep. 4 3683
Google Scholar
-
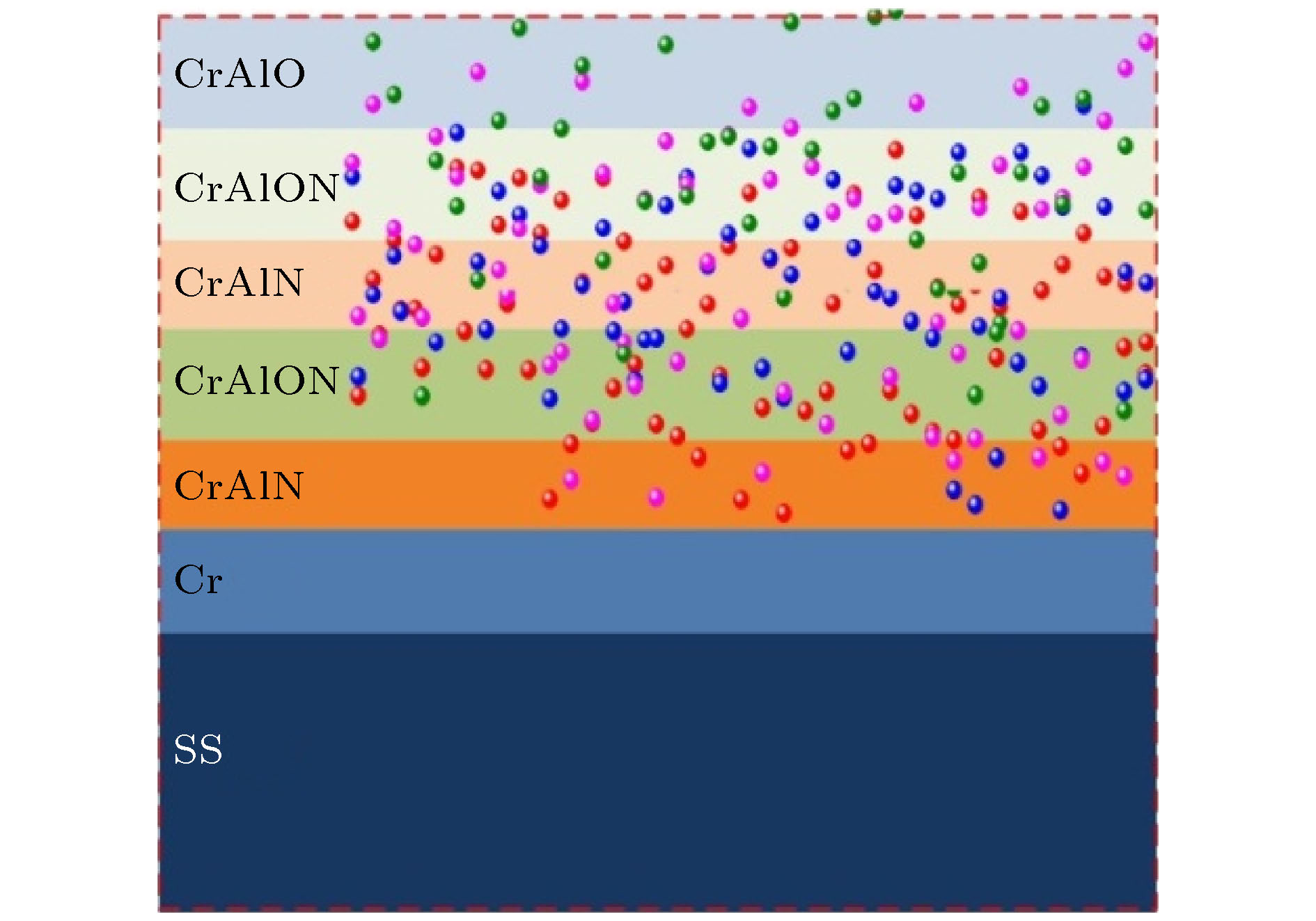
图 1 多吸收层CrAlON基光谱选择性吸收涂层的结构示意图
Fig. 1. Schematic diagram of the multi-absorbing layer CrAlON-based solar selective absorbing coating.

图 2 500 °C高温时效处理过程中多吸收层CrAlON基光谱选择性吸收涂层的反射光谱曲线
Fig. 2. Reflectance spectra of the multi-absorbing layer CrAlON-based solar selective absorbing coatings during the high temperature ageing treatment at 500 °C.

图 3 大气条件下、500 ℃多吸收层CrAlON基光谱选择性吸收涂层不同时效时间后的GIXRD图谱
Fig. 3. GIXRD patterns of the multi-absorbing layer CrAlON-based solar selective absorbing coatings aged at 500 °C for different times in air.

图 4 大气条件下、500 ℃时效220 h后多吸收层CrAlON基光谱选择性吸收涂层的TEM图 (a) 明场像; (b) 选区电子衍射图谱
Fig. 4. TEM images of the multi-absorbing layer CrAlON-based solar selective absorbing coating aged at 500 °C for 220 h in air: (a) Bright-field TEM image; (b) the corresponding selected area electron diffraction (SAED) pattern of the area denoted in Fig. (a)
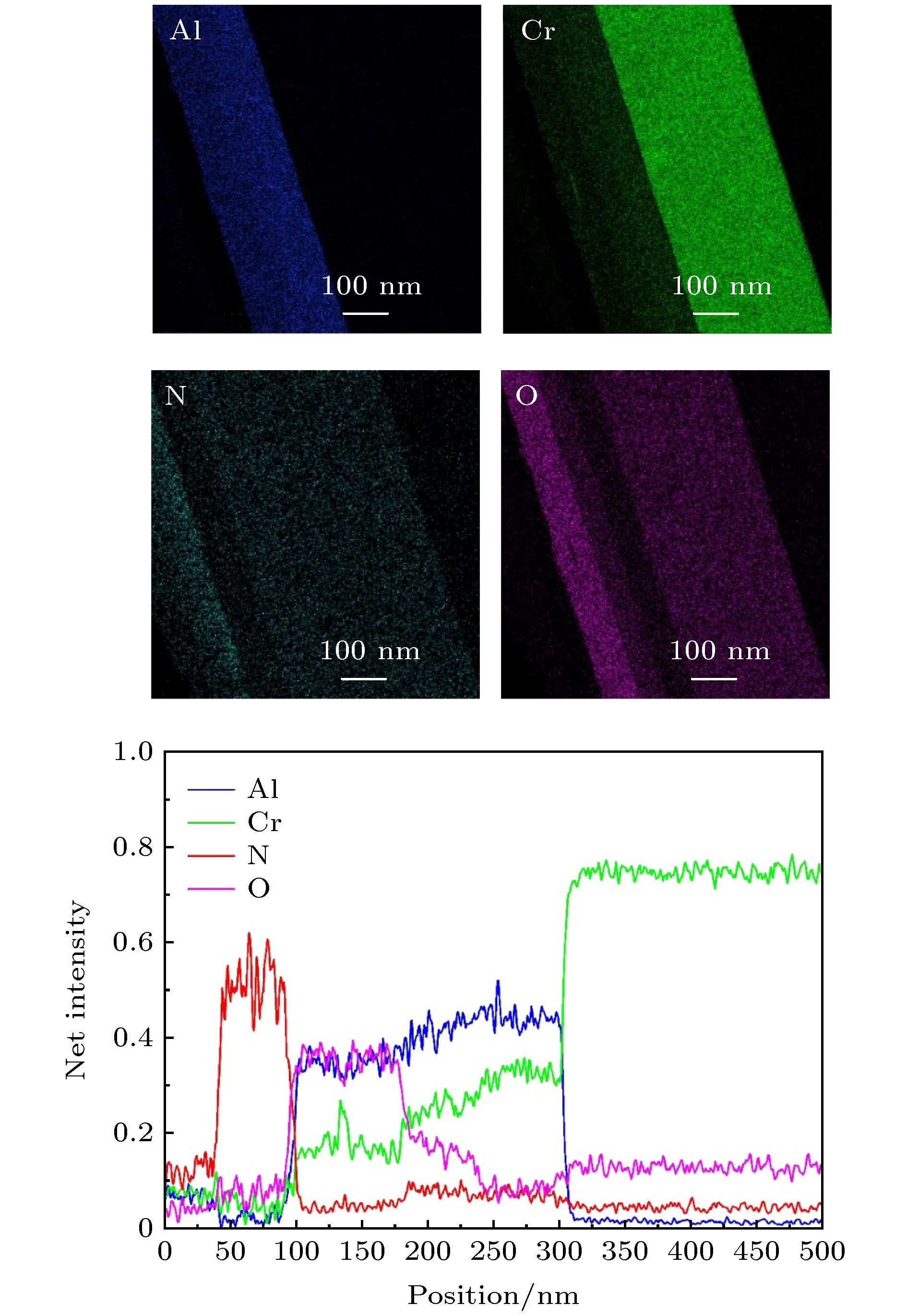
图 5 大气条件下、500 ℃时效220 h后多吸收层CrAlON基光谱选择性吸收涂层截面中Al, Cr, N和O元素的成分分布图和EDS线扫描图
Fig. 5. EDS maps and line scans of the distribution of Al, Cr, N, and O in the multi-absorbing layer CrAlON-based solar selective absorbing coating aged at 500 °C for 220 h in air.

图 6 大气条件下、500 ℃时效220 h后多吸收层CrAlON基光谱选择性吸收涂层的TEM图 (a) 明场像; (b) CrAlO; (c) 外层CrAlON; (d) 外层CrAlN; (e) 内层CrAlON; (f) 内层CrAlN
Fig. 6. TEM images of the multi-absorbing layer CrAlON-based solar selective absorbing coating aged at 500 °C for 220 h in air: (a) The bright-field TEM image; (b) the CrAlO layer; (c) the outer CrAlON layer; (d) the outer CrAlN layer; (e) the inner CrAlON layer; (f) the inner CrAlN layer.

图 7 大气条件下500 ℃时效220 h后多吸收层CrAlON基光谱选择性吸收涂层的HRTEM图 (a) CrAlO减反射层; (b) CrAlON层; (c) CrAlN层
Fig. 7. HRTEM images of the multi-absorbing layer CrAlON-based solar selective absorbing coating aged at 500 °C for 220 h in air: (a) CrAlO layer; (b) CrAlON layer; (c) CrAlN layer.

图 8 大气条件下、500 ℃时效220和1000 h后多吸收层CrAlON基光谱选择性吸收涂层的表面形貌 (a), (b) 220 h; (c), (d) 1000 h
Fig. 8. Morphologies of the multi-absorbing layer CrAlON-based solar selective absorbing coatings aged at 500 °C for 220 and 1000 h in air: (a), (b) Surface morphologies of the coating aged for 220 h; (c), (d) surface morphologies of the coating aged for 1000 h.

图 9 大气条件下、500 ℃时效220 h后多吸收层CrAlON基光谱选择性吸收涂层的AFM形貌
Fig. 9. AFM morphology of the multi-absorbing layer CrAlON-based solar selective absorbing coating after aged at 500 °C for 220 h in air.
表 1 CrAlON基光谱选择性吸收涂层的制备工艺参数
Table 1. Deposition parameters of the CrAlON-based selective absorbing coatings.
Parameters Current/A Ar/sccm O2/sccm N2/sccm Time/ s Cr 90 130 0 0 15 × 60 CrAlN (Inner) 60 100 0 30 60 CrAlON (Inner) 60 120 10 30 60 CrAlN (Outer) 60 100 0 30 60 CrAlON (Outer) 60 120 10 30 60 CrAlO 60 0 130 0 120  下载: 导出CSV
下载: 导出CSV
表 2 500 ℃下时效不同时间后, 多吸收层CrAlON基光谱选择性吸收涂层的吸收率、发射率、选择吸收性α/ε和PC值
Table 2. Absorptance α, emittance ε, selectivity α/ε and PC values of the multi-absorbing layer CrAlON-based solar selective absorbing coatings aged at 500 °C.
Aging parameters α ε α/ε PC As-deposited 0.90 0.15 6 — Aged for 220 h 0.94 0.10 9.4 –0.06 Aged for 1000 h 0.94 0.10 9.4 –0.07
下载: 导出CSV
表 3 高温时效处理220 h多吸收层CrAlON基光谱选择性吸收涂层表面大颗粒的EDS成分(单位: 原子百分比, %)
Table 3. EDS compositions of the macro droplets of CrAlON after aging at 500 °C for 220 h in air (in atomic percent, %).
Position Al Cr O N Site 1 27.22 54.46 15.74 2.58 Site 2 17.46 56.19 20.75 5.61 Site 3 25.33 49.54 21.36 3.78 Average 23.34 53.39 19.28 3.99
下载: 导出CSV
-
[1] 史月艳, 那鸿悦 2009 太阳光谱选择性吸收膜系设计、制备及测评 (第1版) (北京: 清华大学出版社) 第44−65页
Shi Y Y, Na H Y 2009 Design, Preparation and Evaluation of Solar Spectrum Selective absorption Films (1st Ed.) (Beijing: Tsinghua University Press) pp44−65 (in Chinese)
[2] Cao F, McEnaney K, Chen G, Ren Z F 2014 Energy Environ. Sci. 7 1615
Google Scholar
[3] Cao F, Kraemer D, Sun T Y, Lan Y C, Chen G, Ren Z F 2015 Adv. Energy Mater. 5 1401042
Google Scholar
[4] Wang X Y, Gao J H, Hu H B, Zhang H L, Liang L Y, Javaid K, Zhuge F, Cao H T, Wang L 2017 Nano Energy 37 232
Google Scholar
[5] Xue Y F, Wang C, Wang W W, Liu Y, Wu Y X, Ning Y P, Sun Y 2013 Sol. Energy 96 113
Google Scholar
[6] Cheng J S, Wang C, Wang W W, Du X K, Liu Y, Xue Y F, Wang T M, Chen B L 2013 Sol. Energy Mater. Sol. Cells 109 204
Google Scholar
[7] 田广科, 苗树翻, 马天国, 范多旺 2015 太阳能 3 50
Google Scholar
Tian G K, Mi ao, Ma T G, Fan D W 2015 Sol. Energy 3 50
Google Scholar
[8] Ge J P, Zhang Q, Zhang T R, Yin Y D 2008 Angew. Chem. Int. Edit. 47 8924
Google Scholar
[9] Joo S H, Park J Y, Tsung C K, Yamada Y, Yang P, Somorjai G A 2008 Nat. Mater. 8 126
Google Scholar
[10] Gao T, Jelle B P, Gustavsen A J 2013 Nanopart. Res. 15 1370
Google Scholar
[11] Cao A, Veser G 2009 Nat. Mater. 9 75
Google Scholar
[12] Kim T K, VanSaders B, Caldwell E, Shin S, Liu Z, Jin S, Chen R 2016 Sol. Energy 132 257
Google Scholar
[13] Liu H D, Wan Q, Xu Y R, luo C, Chen Y M, Fu D J, Ren F, Luo G, Cheng X D, Hu X J, Yang B 2015 Sol. Energy Mater. Sol. Cells 134 261
Google Scholar
[14] Wu L, Gao J H, Liu Z M, Liang L Y, Xia F, Cao H T 2013 Sol. Energy Mater. Sol. Cells 114 186
Google Scholar
[15] Du M, Hao L, Mi J, Lü F, Liu X P, Jiang L J, Wang S M 2011 Sol. Energy Mater. Sol. Cells 95 1193
Google Scholar
[16] Barshilia H C 2014 Sol. Energy Mater. Sol. Cells 130 322
Google Scholar
[17] Barshilia H C, Selvakumar N, Rajam K S, Biswas A 2008 Sol. Energy Mater. Sol. Cells 92 1425
Google Scholar
[18] Barshilia H C, Selvakumar N, Rajam K S 2007 J. Vac. Sci. Technol., A 25 383
Google Scholar
[19] 史月艳, 那鸿悦 2009 太阳光谱选择性吸收膜系设计、制备及测评 (第1版) (北京: 清华大学出版社) 第67−147页
Shi Y Y, Na H Y 2009 Design, Preparation and Evaluation of Solar Spectrum Selective absorption Films (1st Ed.) (Beijing: Tsinghua University Press) pp67−147(in Chinese)
[20] Zou C W, Xie W, Shao L X 2016 Sol. Energy Mater. Sol. Cells 153 9
Google Scholar
[21] Liu H D, Fu T R, Duan M H, Wan Q, luo C, Chen Y M, Fu D J, Ren F, Li Q Y, Cheng X D, Yang B, Hu X J 2016 Sol. Energy Mater. Sol. Cells 157 108
Google Scholar
[22] Gong D Q, Liu H D, Luo G, Zhang P, Cheng X D, Yang B, Wang Y B, Min J, Wang W X, Chen S P, Cui Z Q, Li K W, Hu L F 2015 Sol. Energy Mater. Sol. Cells 136 167
Google Scholar
[23] Gammer C, Mangler C, Rentenberger C, Karnthaler H P 2010 Scr. Mater. 63 312
Google Scholar
[24] Gao X H, Guo Z M, Geng Q F, Ma P J, Wang A Q, Liu G 2016 Sol. Energy 140 199
Google Scholar
[25] van den Oetelaar L C A, Nooij O W, Oerlemans S, Denier van der Gon A W, Brongersma H H, Lefferts L, Roosenbrand A G, van Veen J A R 1998 J. Phys. Chem. B 102 3445
Google Scholar
[26] Malis O, Radu M, Mott D, Wanjala B, Luo J, Zhong C J 2009 Nanotechnology 20 245708
Google Scholar
[27] Liao H, Fisher A, Xu Z J 2015 Small 11 3221
Google Scholar
[28] Dean J A 1990 Mater. Manuf. Processes 5 687
Google Scholar
[29] Clark B G, Hattar K, Marshall M T, Chookajorn T, Boyce B L, Schuh C A 2016 JOM 68 1625
Google Scholar
[30] Han L L, Meng Q P, Wang D L, Zhu Y M, Wang J, Du X W, Stach E A, Xin H L 2016 Nat. Commun. 7 13335
Google Scholar
[31] Huolin L X, Selim A, Runzhe T, Arda G, Chong-Min W, Libor K, Eric A S, Lin-Wang W, Miquel S, Gabor A S, Haimei Z 2014 Nano Lett. 14 3203
Google Scholar
[32] Wang C M, Genc A, Cheng H, Pullan L, Baer D R, Bruemmer S M 2014 Sci. Rep. 4 3683
Google Scholar

 下载:
下载:
计量
- 文章访问数: 10775
- PDF下载量: 118
- 被引次数: 0
