










-
本文在第一性原理计算基础上结合非平衡格林函数方法, 研究了量子干涉效应对连接镍电极的二噻吩硼烷(dithienoborepin, DTB)分子结自旋输运性质的影响, 并通过氨基和硝基钝化实现了对二噻吩硼烷分子异构体(DTB-A和DTB-B)的区分. 结果表明, 原始的DTB-A和DTB-B分子结在费米能级两侧都有一个自旋向上透射峰和一个自旋向下透射峰, 且两个透射峰的能量位置和高度基本相同. 因此, 原始DTB-A和DTB-B分子结的自旋向上和自旋向下电流曲线基本重合, 不能被明显区分. 然而, 研究发现量子干涉效应能不同程度地增强氨基钝化DTB-A分子结费米能级两侧分子轨道的自旋极化输运能力, 并减弱氨基钝化DTB-B分子结费米能级两侧分子轨道的自旋极化输运能力. 此外, 研究还发现量子干涉效应可以显著提高硝基钝化DTB-B分子结费米能级两侧分子轨道的自旋极化输运能力, 同时减弱硝基钝化DTB-A分子结费米能级两侧分子轨道的自旋极化输运能力. 由于量子干涉效应对氨基和硝基钝化的DTB异构体分子结自旋输运能力有不同的调制作用, 因此可以通过测量氨基和硝基钝化分子结的自旋电流值来区分DTB分子的两种异构体.Previous research results show that the conductance difference in molecular junction caused by quantum interference (QI) effect is an important way to identify isomers or improve the recognition sensitivity. Recently, single-molecule conductance of two fully π-conjugated dithienoborepin (DTB) isomers (DTB-A and DTB-B) with tricoordinate boron centers has been measured by using the scanning tunneling microscopy break junction technique. The result shows that QI can enhance chemical responsivity in single-molecule DTB junction. In this work, the first-principles method based on density functional theory and non-equilibrium Green's function is used to study the influence of QI effect on spin-transport property of DTB molecular junction connected to the nickel electrode, and the purpose of distinguishing DTB isomers (DTB-A and DTB-B) is realized by using amino and nitro passivation. The results show that the pristine DTB-A molecule and DTB-B molecule both have a up-spin transmission peak dominated by HOMO and a down-spin transmission peak dominated by LUMO on both sides of the Fermi level, and the energy positions and coefficients of two transmission peaks are basically the same. Therefore, the up-spin and down-spin current curves of the two junctions basically coincide, so that it is impossible to clearly distinguish the two isomers of DTB molecule simply by spin current. The QI can enhance the spin-polarized transport capability of two orbitals of amino-passivated DTB-A molecule to varying degrees but weaken the spin-polarized transport capability of two orbitals of amino-passivated DTB-B molecule. Therefore, the current of DTB-A molecular junction passivated by amino group is always higher than that of DTB-B molecular junction passivated by amino group. However, the QI can greatly enhance the spin-polarized transport capability of two orbitals of nitro-passivated DTB-B molecule but weaken the spin-polarized transport capability of two orbitals of nitro-passivated DTB-A molecule. Therefore, the current of DTB-B molecular junction passivated by nitro is always higher than that of DTB-A molecular junction passivated by nitro. Because the QI has different effects on the spin-transport capability of DTB-A and DTB-B passivated by amino or nitro group, so the two isomers of DTB molecule can be distinguished by measuring the spin current value. The above conclusions provide more theoretical guidance for the practical preparation of spin molecular junctions and the regulation of their spin-transport performance in the future.
-
Keywords:
- first principles /
- density functional theory /
- molecular device /
- quantum interference /
- spin-transport
[1] Aviram A, Ratner M A 1974 Chem. Phys. Lett. 29 277
 Google Scholar
Google Scholar
[2] Perrin M L, Frisenda R, Koole M, Seldenthuis J S, Gil J A C, Valkenier H, Hummelen J C, Renaud N, Grozema F C, Thijssen J M, Dulić D, van der Zant H S J 2014 Nat. Nanotechnol. 9 830
Google Scholar
[3] Koley S, Chakrabarti S 2018 Chem. Eur. J. 24 5876
Google Scholar
[4] Sharma P, Bernard L S, Bazigos A, Magrez A, Ionescu A M 2015 ACS Nano 9 620
Google Scholar
[5] Kumar S, Wang Z, Davila N, Kumari N, Norris K J, Huang X, Strachan J P, Vine D, Kilcoyne A L D, Nishi Y, Williams R S 2017 Nat. Commun. 8 658
Google Scholar
[6] Li Z L, Sun F, Bi J J, Liu R, Suo Y Q, Fu H Y, Zhang G P, Song Y Z, Wang D, Wang C K 2019 Physica E 106 270
Google Scholar
[7] Liu Q, Li J J, Wu D, Deng X Q, Zhang Z H, Fan Z Q, Chen K Q 2021 Phys. Rev. B 104 045412
Google Scholar
[8] Komeda J, Takada K, Maeda H, Fukui N, Tsuji T, Nishihara H 2022 Chem. Eur. J. 28 e202201316
Google Scholar
[9] Zhao J, Zeng H, Wang D, Yao G 2020 Appl. Surf. Sci. 519 146203
Google Scholar
[10] Petersen M Å, Rasmussen B, Andersen N N, Sauer S P A, Nielsen M B, Beeren S R, Pittelkow M 2017 Chem. Eur. J. 23 17010
Google Scholar
[11] Fan Z Q, Zhang Z H, Yang S Y 2020 Nanoscale 12 21750
Google Scholar
[12] Jiang J, Kula M, Lu W, Luo Y 2005 Nano Lett. 5 1551
Google Scholar
[13] Lambert C J 2015 Chem. Soc. Rev. 44 875
Google Scholar
[14] Gehring P, Thijssen J M, van der Zant H S J 2019 Nat. Rev. Phys. 1 381
Google Scholar
[15] Manrique D Z, Huang C, Baghernejad M, Zhao X, Al-Owaedi O A, Sadeghi H, Kaliginedi V, Hong W, Gulcur M, Wandlowski T, Bryce M R, Lambert C J 2015 Nat. Commun. 6 6389
Google Scholar
[16] Fan Z Q, Sun W Y, Zhang Z H, Deng X Q, Tang G P, Xie H Q 2017 Carbon 122 687
Google Scholar
[17] Li Z L, Bi J J, Liu R, Yi X H, Fu H Y, Sun F, Wei M Z, Wang C K 2017 Chin. Phys. B 26 098508
Google Scholar
[18] Yang Y, Gantenbein M, Alqorashi A, Wei J, Sangtarash S, Hu D, Sadeghi H, Zhang R, Pi J, Chen L, Huang X, Li R, Liu J, Shi J, Hong W, Lambert C J, Bryce M R 2018 J. Phys. Chem. C 122 14965
Google Scholar
[19] Liu X, Sangtarash S, Reber D, Zhang D, Sadeghi H, Shi J, Xiao Z Y, Hong W, Lambert C J, Liu S X 2017 Angew. Chem. 129 179
Google Scholar
[20] Borges A, Fung E D, Ng F, Venkataraman L, Solomon G C 2016 J. Phys. Chem. Lett. 7 4825
Google Scholar
[21] Frisenda R, Janssen V A E C, Grozema F C, van der Zant H S J, Renaud N 2016 Nat. Chem. 8 1099
Google Scholar
[22] Liu R, Bi J J, Xie Z, Yin K, Wang D, Zhang G P, Xiang D, Wang C K, Li Z L 2018 Phys. Rev. Appl. 9 054023
Google Scholar
[23] Zhang Y P, Chen L C, Zhang Z Q, Cao J J, Tang C, Liu J, Duan L L, Huo Y, Shao X, Hong W, Zhang H L 2018 J. Am. Chem. Soc. 140 6531
Google Scholar
[24] Yang G, Sangtarash S, Liu Z, Li X, Sadeghi H, Tan Z, Li R, Zheng J, Dong X, Liu J, Yang Y, Shi J, Xiao Z, Zhang G, Lambert C, Hong W, Zhang D 2017 Chem. Sci. 8 7505
Google Scholar
[25] Li Y, Buerkle M, Li G, Rostamian A, Wang H, Wang Z, Bowler D R, Miyazaki T, Xiang L, Asai Y, Zhou G, Tao N 2019 Nat. Mater. 18 357
Google Scholar
[26] Huang B, Liu X, Yuan Y, Hong Z W, Zheng J F, Pei L Q, Shao Y, Li J F, Zhou X S, Chen J Z, Jin S, Mao B W 2018 J. Am. Chem. Soc. 140 17685
Google Scholar
[27] Bai J, Daaoub A, Sangtarash S, Li X, Tang Y, Zou Q, Sadeghi H, Liu S, Huang X, Tan Z, Liu J, Yang Y, Shi J, Mészáros G, Chen W, Lambert C, Hong W 2019 Nat. Mater. 18 364
Google Scholar
[28] Zheng J, Liu J, Zhuo Y, Li R, Jin X, Yang Y, Chen Z B, Shi J, Xiao Z, Hong W, Tian Z Q 2018 Chem. Sci. 9 5033
Google Scholar
[29] Liu J, Zhao X, Al-Galiby Q, Huang X, Zheng J, Li R, Huang C, Yang Y, Shi J, Manrique D Z, Lambert C J, Bryce M R, Hong W 2017 Angew. Chem. 129 13241
Google Scholar
[30] Cai S, Deng W, Huang F, Chen L, Tang C, He W, Long S, Li R, Tan Z, Liu J, Shi J, Liu Z, Xiao Z, Zhang D, Hong W 2019 Angew. Chem. Int. Ed. 58 3829
Google Scholar
[31] Ozawa H, Baghernejad M, Al-Owaedi O A, Kaliginedi V, Nagashima T, Ferrer J, Wandlowski T, García-Suárez V M, Broekmann P, Lambert C J, Haga M A 2016 Chem. Eur. J. 22 12732
Google Scholar
[32] Greenwald J E, Cameron J, Findlay N J, Fu T, Gunasekaran S, Skabara P J, Venkataraman L 2021 Nat. Nanotechnol. 16 313
Google Scholar
[33] Trasobares J, Vuillaume D, Théron D, Clément N 2016 Nat. Commun. 7 12850
Google Scholar
[34] Niu L L, Fu H Y, Suo Y Q, Liu R, Sun F, Wang S S, Zhang G P, Wang C K, Li Z L 2021 Phys. E 128 114542
Google Scholar
[35] Wu D, Huang L, Jia P Z, Cao X H, Fan Z Q, Zhou W X, Chen K Q 2021 Appl. Phys. Lett. 119 063503
Google Scholar
[36] Fan Z Q, Xie F, Jiang X W, Wei Z, Li S S 2016 Carbon 110 200
Google Scholar
[37] Kushmerick J 2009 Nature 462 994
Google Scholar
[38] Richter S, Mentovich E, Elnathan R 2018 Adv. Mater. 30 1706941
Google Scholar
[39] Liu R, Han Y, Sun F, Khatri G, Kwon J, Nickle C, Wang L, Wang C K, Thompson D, Li Z L, Nijhuis C A, del Barco E 2022 Adv. Mater. 34 2202135
Google Scholar
[40] Fan Z Q, Sun W Y, Jiang X W, Zhang Z H, Deng X Q, Tang G P, Xie H Q, Long M Q 2017 Carbon 113 18
Google Scholar
[41] Zhou Q, Yang Y, Ni J, Li Z, Zhang Z 2010 Nano Res. 3 423
Google Scholar
[42] Baghernejad M, Van Dyck C, Bergfield J, Levine D R, Gubicza A, Tovar J D, Calame M, Broekmann P, Hong W 2019 Chem. Eur. J. 25 15141
Google Scholar
[43] Büttiker M, Imry Y, Landauer R, Pinhas S 1985 Phys. Rev. B 31 6207
Google Scholar
[44] Smidstrup S, Markussen T, Vancraeyveld P, Wellendorff J, Schneider J, Gunst T, Verstichel B, Stradi D, Khomyakov P A, Vej-Hansen U G, Lee M E, Chill S T, Rasmussen F, Penazzi G, Corsetti F, Ojanperä A, Jensen K, Palsgaard M L N, Martinez U, Blom A, Brandbyge M, Stokbro K 2019 J. Phys. Condens. Matter 32 015901
Google Scholar
[45] Pan H, Tang L M, Chen K Q 2022 Phys. Rev. B 105 064401
Google Scholar
-
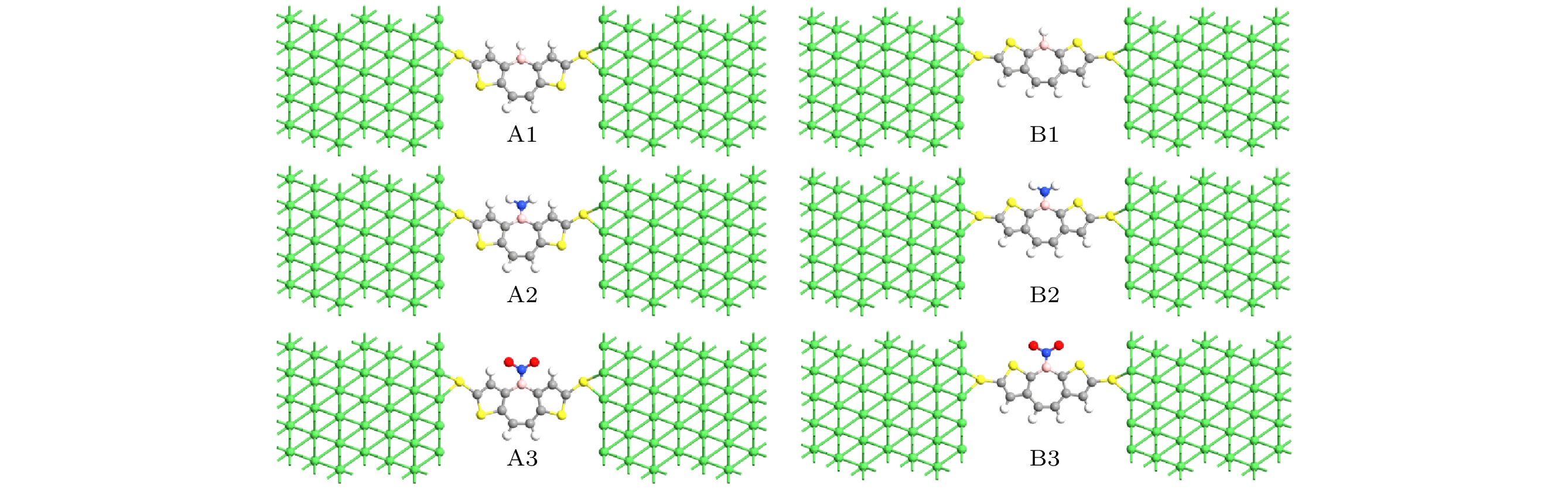
图 1 氨基或硝基钝化前后DTB-A和DTB-B分子结示意图
Fig. 1. Schematic diagrams of DTB-A and DTB-B molecular junctions before and after amino or nitro passivation.
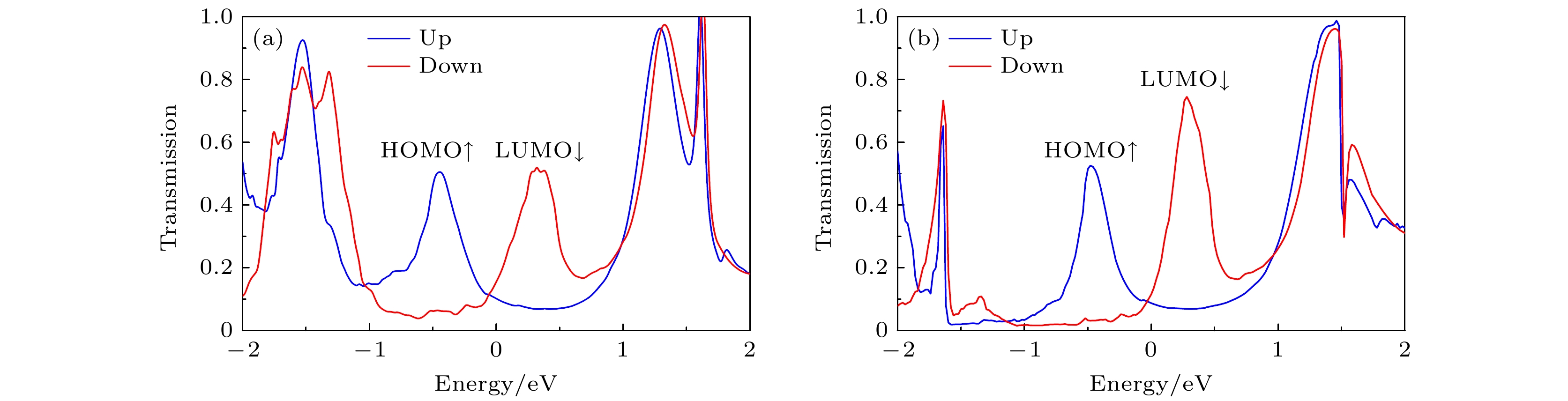
图 2 零偏压下(a) A1和(b) B1的自旋透射谱, 蓝线和红线分别代表自旋向上和自旋向下, 费米能级在能量尺度上被设定为零
Fig. 2. Spin-transmission spectra of (a) A1 and (b) B1 at zero bias, the blue line and the red line represent up-spin and down-spin, respectively. The Fermi level is set at zero in the energy scale.

图 3 A1的(a) HOMO↑对应的透射本征态和(b) LUMO↓对应的透射本征态; B1的(c) HOMO↑对应的透射本征态和(d) LUMO↓对应的透射本征态
Fig. 3. The transmission eigenstates of (a) HOMO↑ and (b) LUMO↓of A1; the transmission eigenstates of (c) HOMO↑and (d) LUMO↓ of B1.
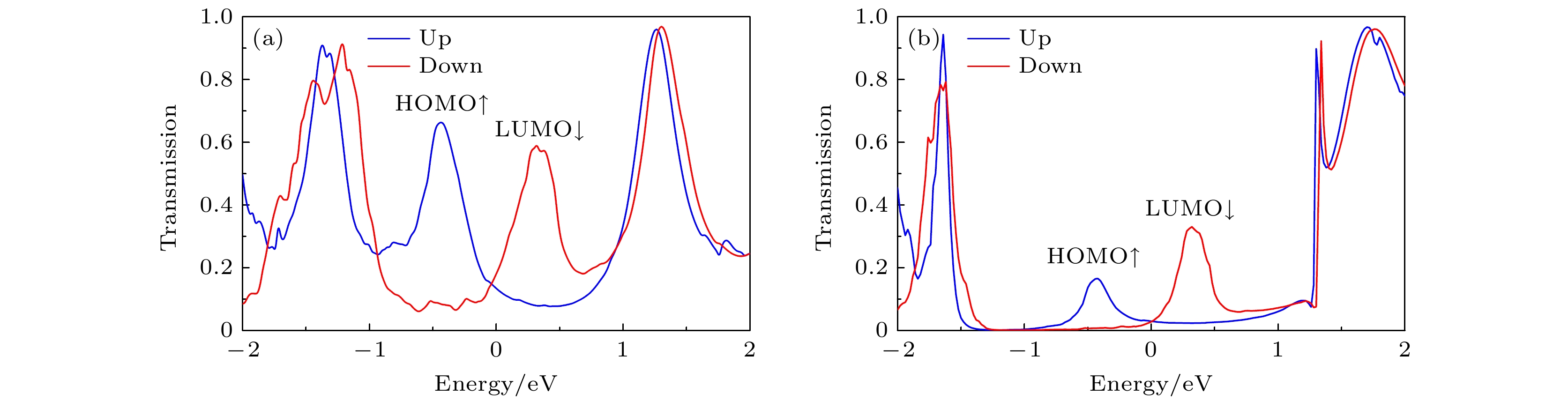
图 4 零偏压下(a) A2和(b) B2的自旋透射谱, 蓝线和红线分别代表自旋向上和自旋向下, 费米能级在能量尺度上被设定为零
Fig. 4. Spin-transport spectra of (a) A2 and (b) B2 at zero bias, the blue line and the red line represent up-spin and down-spin, respectively. The Fermi level is set at zero in the energy scale.
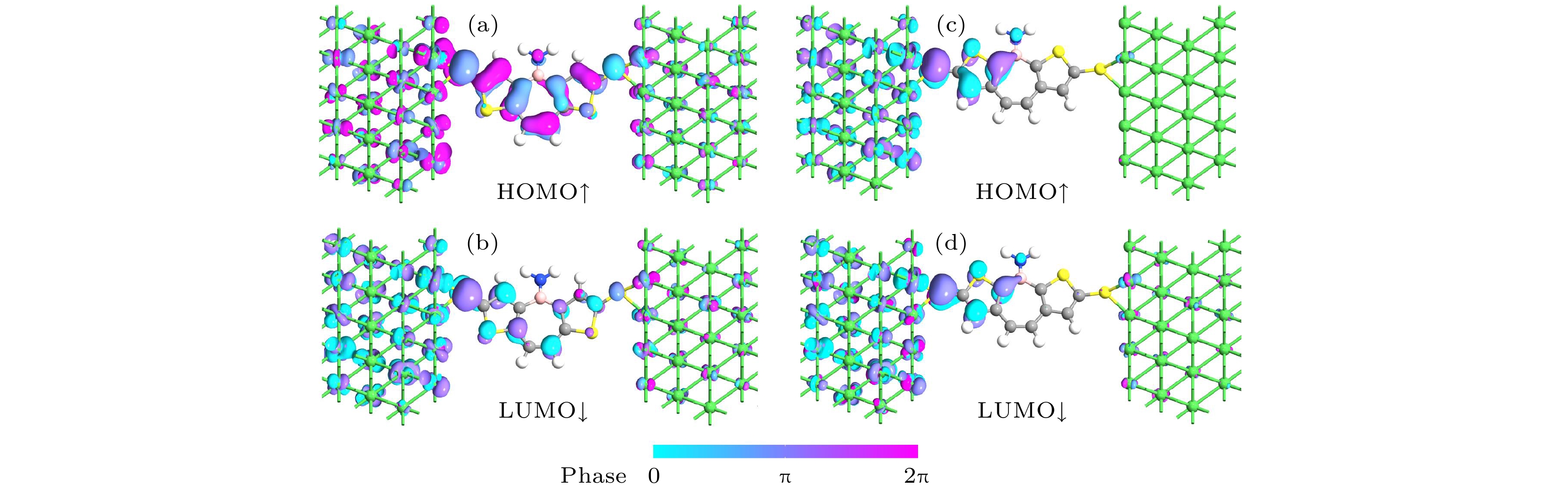
图 5 A2的(a) HOMO↑对应的透射本征态和(b) LUMO↓对应的透射本征态; B2的(c) HOMO↑对应的透射本征态和(d) LUMO↓对应的透射本征态
Fig. 5. The transmission eigenstates of (a) up-spin HOMO and (b) down-spin LUMO of A2; the transmission eigenstates of (c) up-spin HOMO and (d) down-spin LUMO of B2.
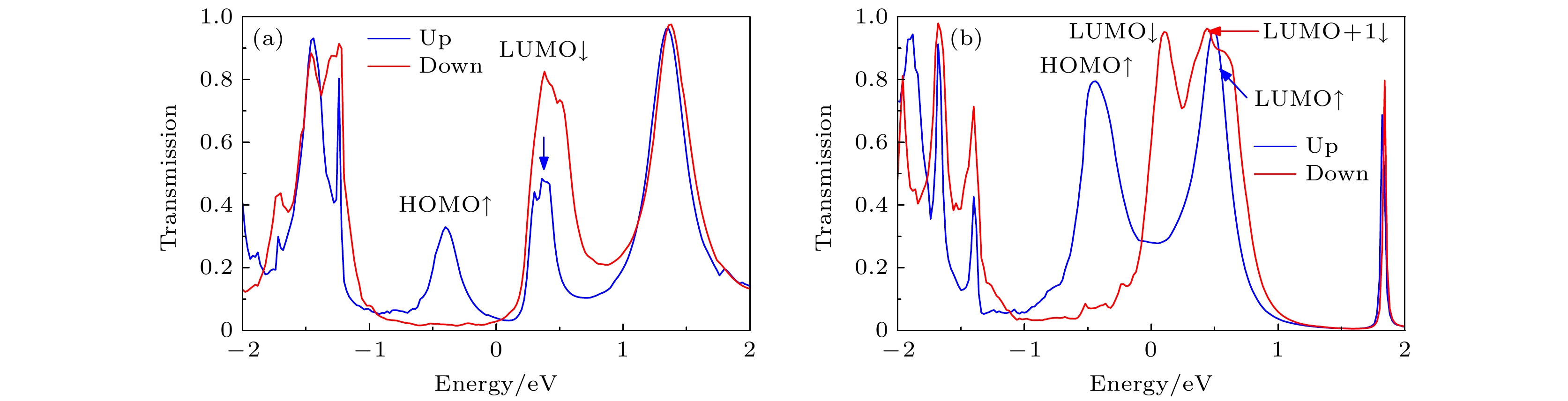
图 6 零偏压下(a) A3和(b) B3的自旋透射谱, 蓝线和红线分别代表自旋向上和自旋向下, 费米能级在能量尺度被设定为零
Fig. 6. Spin-transport spectra of (a) A3 and (b) B3 at zero bias, the blue line and the red line represent up-spin and down-spin, respectively. The Fermi level is set at zero in the energy scale.

图 7 A3的(a) HOMO↑和(b) LUMO↓对应的透射本征态; B3的(c)HOMO↑和(d)LUMO↓对应的透射本征态
Fig. 7. The transmission eigenstates of (a) up-spin HOMO and (b) down-spin LUMO of A3; the transmission eigenstates of (c) up-spin HOMO and (d) down-spin LUMO of B3.
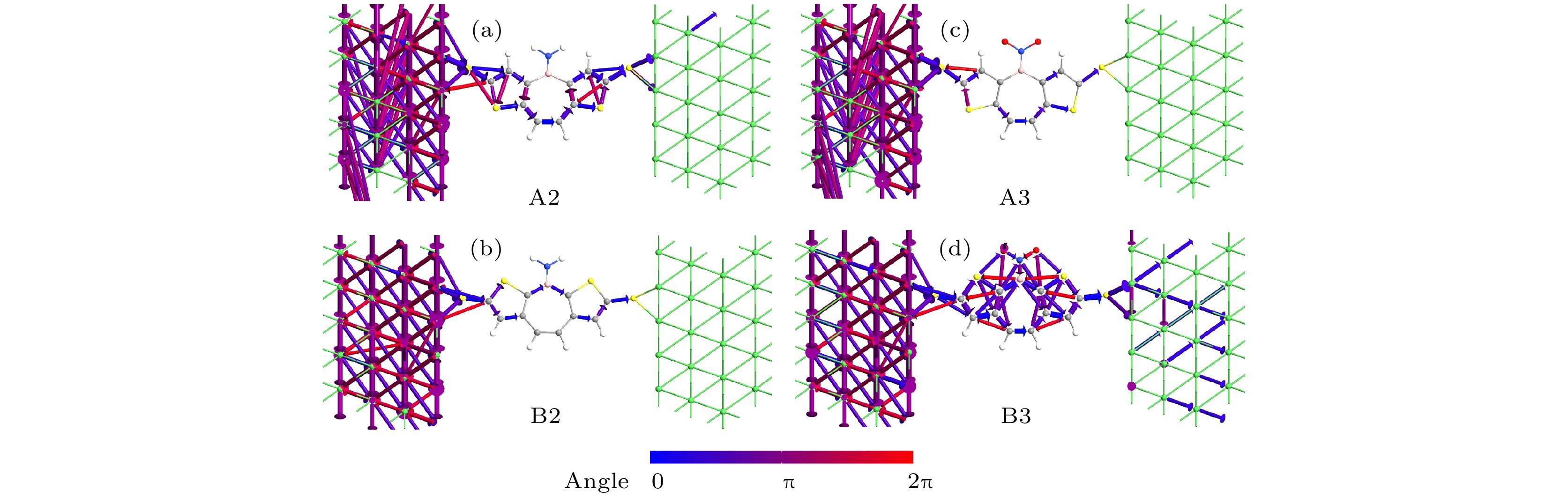
图 8 零偏压下A2, B2, A3和B3在费米能级上的传输路径
Fig. 8. The transmission pathways of A2, B2, A3 and B3 at Fermi level under zero bias.
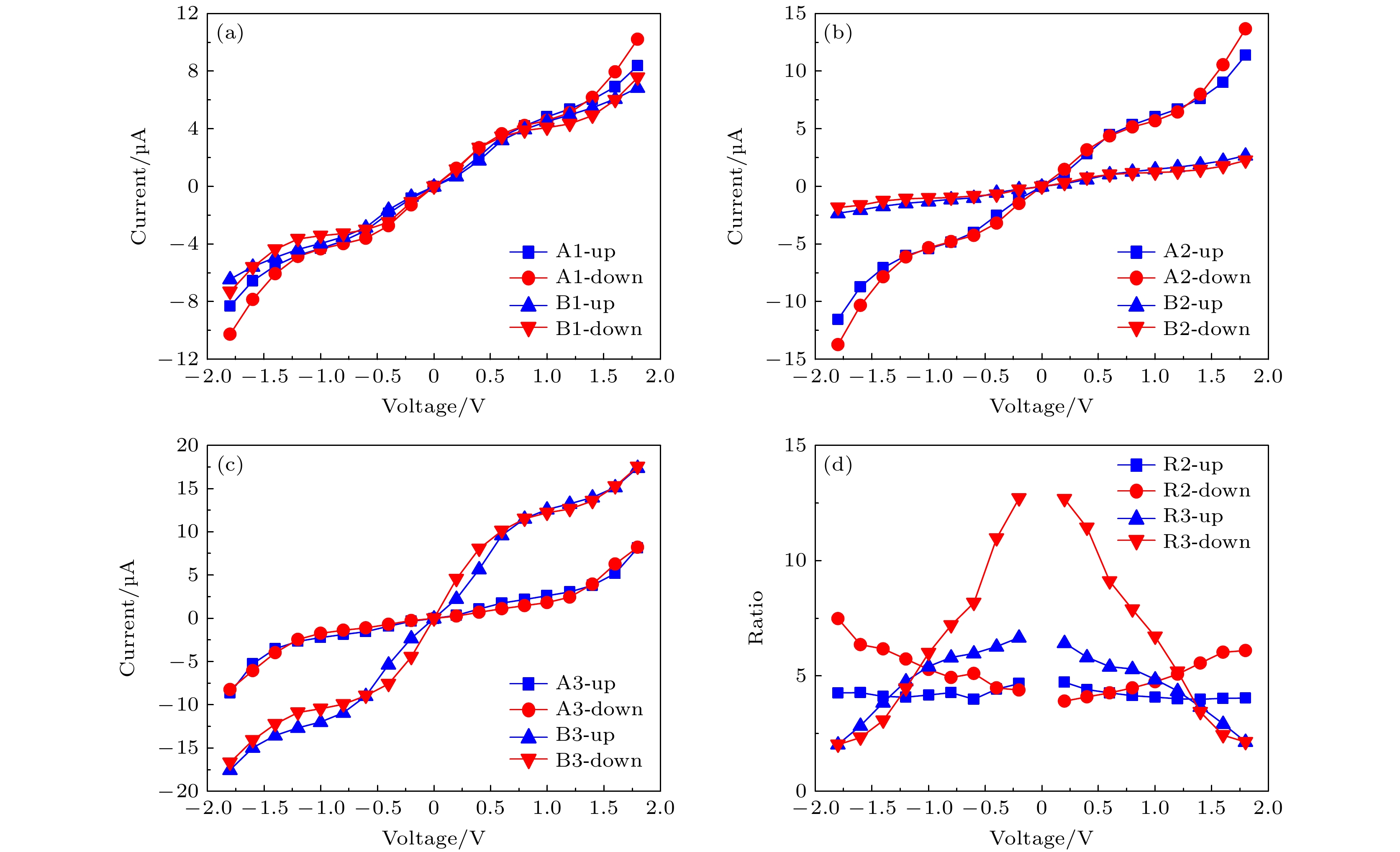
图 9 (a)—(c) A1, B1, A2, B2, A3和B3分别在±1.8 V范围内的自旋电流-电压特性; (d)在同一自旋状态下, A2与B2和A3与B3随电压的变化的自旋电流之比
Fig. 9. (a)–(c) Spin-resolved current-voltage characteristics of A1, B1, A2, B2, A3, and B3 in the range of ±1.8 V, respectively; (d) the variation of spin-current ratios with voltages in the same spin state of A2 to B2 and A3 to B3.

图 10 (a) A1和(b) B1在0 V, ±0.8 V 和 ±1.6 V偏压下的自旋透射谱, 蓝线和红线分别代表自旋向上和自旋向下, 黑色实线之间的区域为偏压窗口, 费米能级在能量尺度上被设定为零
Fig. 10. Spin-transmission spectra of (a) A1 and (b) B1 at 0 V, ±0.8 V 和 ±1.6 V. The blue line and the red line represent up-spin and down-spin, respectively. The region between the black solid lines is the bias window. The Fermi level is set at zero in the energy scale.
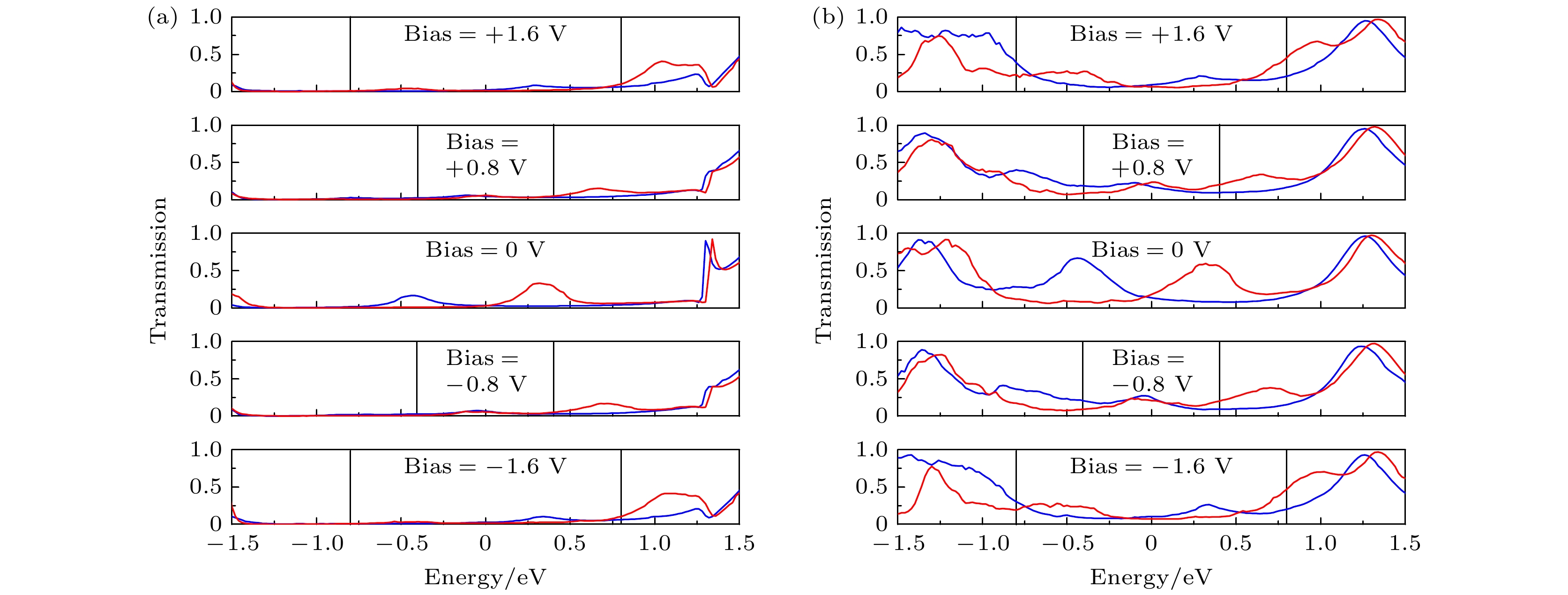
图 11 (a) A2和(b) B2在0 V, ±0.8 V 和 ±1.6 V偏压下的自旋透射谱, 蓝线和红线分别代表自旋向上和自旋向下, 黑色实线之间的区域为偏压窗口, 费米能级在能量尺度上被设定为零
Fig. 11. Spin-transmission spectra of (a) A2 and (b) B2 at 0 V, ±0.8 V 和 ±1.6 V. The blue line and the red line represent up-spin and down-spin, respectively. The region between the black solid lines is the bias window. The Fermi level is set at zero in the energy scale.
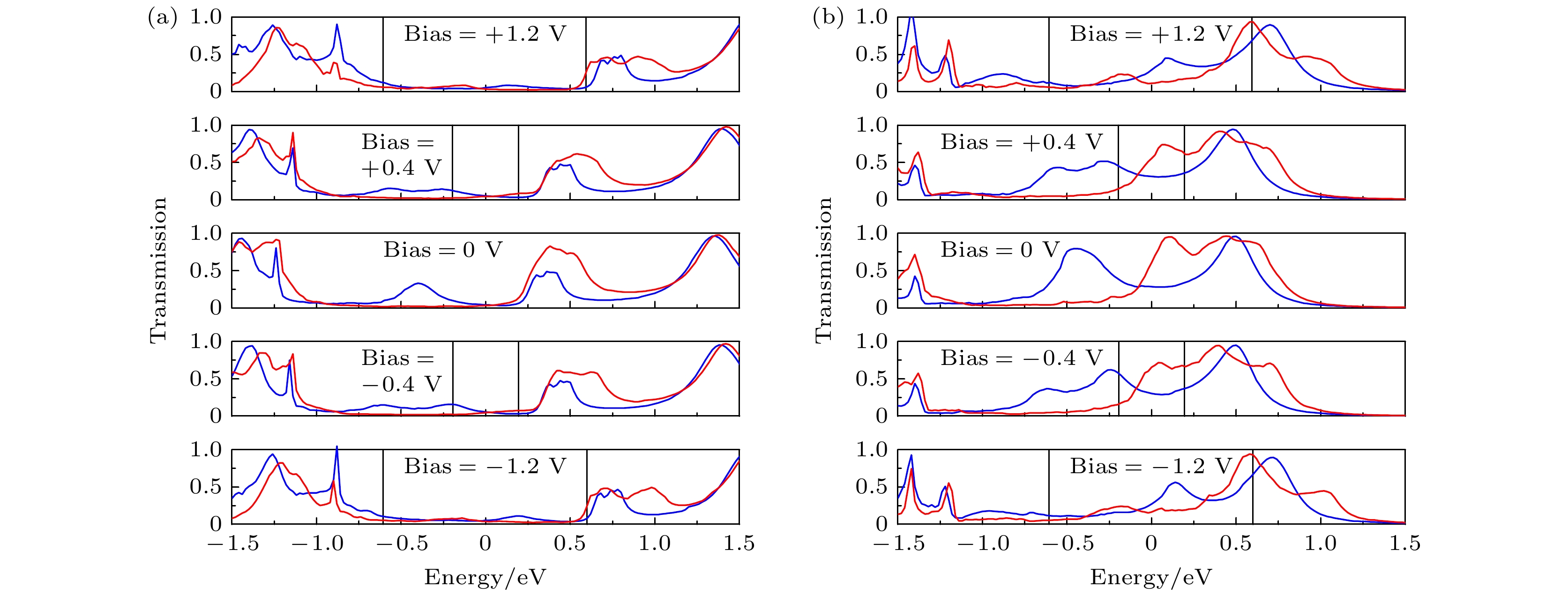
图 12 (a) A3和(b) B3在0 V, ±0.4 V 和 ±1.2 V偏压下的自旋透射谱, 蓝线和红线分别代表自旋向上和自旋向下, 黑色实线之间的区域为偏压窗口, 费米能级在能量尺度上被设定为零
Fig. 12. Spin-transmission spectra of (a) A3 and (b) B3 at 0 V, ±0.4 V 和 ±1.2 V. The blue line and the red line represent up-spin and down-spin, respectively. The region between the black solid lines is the bias window. The Fermi level is set at zero in the energy scale.
-
[1] Aviram A, Ratner M A 1974 Chem. Phys. Lett. 29 277
Google Scholar
[2] Perrin M L, Frisenda R, Koole M, Seldenthuis J S, Gil J A C, Valkenier H, Hummelen J C, Renaud N, Grozema F C, Thijssen J M, Dulić D, van der Zant H S J 2014 Nat. Nanotechnol. 9 830
Google Scholar
[3] Koley S, Chakrabarti S 2018 Chem. Eur. J. 24 5876
Google Scholar
[4] Sharma P, Bernard L S, Bazigos A, Magrez A, Ionescu A M 2015 ACS Nano 9 620
Google Scholar
[5] Kumar S, Wang Z, Davila N, Kumari N, Norris K J, Huang X, Strachan J P, Vine D, Kilcoyne A L D, Nishi Y, Williams R S 2017 Nat. Commun. 8 658
Google Scholar
[6] Li Z L, Sun F, Bi J J, Liu R, Suo Y Q, Fu H Y, Zhang G P, Song Y Z, Wang D, Wang C K 2019 Physica E 106 270
Google Scholar
[7] Liu Q, Li J J, Wu D, Deng X Q, Zhang Z H, Fan Z Q, Chen K Q 2021 Phys. Rev. B 104 045412
Google Scholar
[8] Komeda J, Takada K, Maeda H, Fukui N, Tsuji T, Nishihara H 2022 Chem. Eur. J. 28 e202201316
Google Scholar
[9] Zhao J, Zeng H, Wang D, Yao G 2020 Appl. Surf. Sci. 519 146203
Google Scholar
[10] Petersen M Å, Rasmussen B, Andersen N N, Sauer S P A, Nielsen M B, Beeren S R, Pittelkow M 2017 Chem. Eur. J. 23 17010
Google Scholar
[11] Fan Z Q, Zhang Z H, Yang S Y 2020 Nanoscale 12 21750
Google Scholar
[12] Jiang J, Kula M, Lu W, Luo Y 2005 Nano Lett. 5 1551
Google Scholar
[13] Lambert C J 2015 Chem. Soc. Rev. 44 875
Google Scholar
[14] Gehring P, Thijssen J M, van der Zant H S J 2019 Nat. Rev. Phys. 1 381
Google Scholar
[15] Manrique D Z, Huang C, Baghernejad M, Zhao X, Al-Owaedi O A, Sadeghi H, Kaliginedi V, Hong W, Gulcur M, Wandlowski T, Bryce M R, Lambert C J 2015 Nat. Commun. 6 6389
Google Scholar
[16] Fan Z Q, Sun W Y, Zhang Z H, Deng X Q, Tang G P, Xie H Q 2017 Carbon 122 687
Google Scholar
[17] Li Z L, Bi J J, Liu R, Yi X H, Fu H Y, Sun F, Wei M Z, Wang C K 2017 Chin. Phys. B 26 098508
Google Scholar
[18] Yang Y, Gantenbein M, Alqorashi A, Wei J, Sangtarash S, Hu D, Sadeghi H, Zhang R, Pi J, Chen L, Huang X, Li R, Liu J, Shi J, Hong W, Lambert C J, Bryce M R 2018 J. Phys. Chem. C 122 14965
Google Scholar
[19] Liu X, Sangtarash S, Reber D, Zhang D, Sadeghi H, Shi J, Xiao Z Y, Hong W, Lambert C J, Liu S X 2017 Angew. Chem. 129 179
Google Scholar
[20] Borges A, Fung E D, Ng F, Venkataraman L, Solomon G C 2016 J. Phys. Chem. Lett. 7 4825
Google Scholar
[21] Frisenda R, Janssen V A E C, Grozema F C, van der Zant H S J, Renaud N 2016 Nat. Chem. 8 1099
Google Scholar
[22] Liu R, Bi J J, Xie Z, Yin K, Wang D, Zhang G P, Xiang D, Wang C K, Li Z L 2018 Phys. Rev. Appl. 9 054023
Google Scholar
[23] Zhang Y P, Chen L C, Zhang Z Q, Cao J J, Tang C, Liu J, Duan L L, Huo Y, Shao X, Hong W, Zhang H L 2018 J. Am. Chem. Soc. 140 6531
Google Scholar
[24] Yang G, Sangtarash S, Liu Z, Li X, Sadeghi H, Tan Z, Li R, Zheng J, Dong X, Liu J, Yang Y, Shi J, Xiao Z, Zhang G, Lambert C, Hong W, Zhang D 2017 Chem. Sci. 8 7505
Google Scholar
[25] Li Y, Buerkle M, Li G, Rostamian A, Wang H, Wang Z, Bowler D R, Miyazaki T, Xiang L, Asai Y, Zhou G, Tao N 2019 Nat. Mater. 18 357
Google Scholar
[26] Huang B, Liu X, Yuan Y, Hong Z W, Zheng J F, Pei L Q, Shao Y, Li J F, Zhou X S, Chen J Z, Jin S, Mao B W 2018 J. Am. Chem. Soc. 140 17685
Google Scholar
[27] Bai J, Daaoub A, Sangtarash S, Li X, Tang Y, Zou Q, Sadeghi H, Liu S, Huang X, Tan Z, Liu J, Yang Y, Shi J, Mészáros G, Chen W, Lambert C, Hong W 2019 Nat. Mater. 18 364
Google Scholar
[28] Zheng J, Liu J, Zhuo Y, Li R, Jin X, Yang Y, Chen Z B, Shi J, Xiao Z, Hong W, Tian Z Q 2018 Chem. Sci. 9 5033
Google Scholar
[29] Liu J, Zhao X, Al-Galiby Q, Huang X, Zheng J, Li R, Huang C, Yang Y, Shi J, Manrique D Z, Lambert C J, Bryce M R, Hong W 2017 Angew. Chem. 129 13241
Google Scholar
[30] Cai S, Deng W, Huang F, Chen L, Tang C, He W, Long S, Li R, Tan Z, Liu J, Shi J, Liu Z, Xiao Z, Zhang D, Hong W 2019 Angew. Chem. Int. Ed. 58 3829
Google Scholar
[31] Ozawa H, Baghernejad M, Al-Owaedi O A, Kaliginedi V, Nagashima T, Ferrer J, Wandlowski T, García-Suárez V M, Broekmann P, Lambert C J, Haga M A 2016 Chem. Eur. J. 22 12732
Google Scholar
[32] Greenwald J E, Cameron J, Findlay N J, Fu T, Gunasekaran S, Skabara P J, Venkataraman L 2021 Nat. Nanotechnol. 16 313
Google Scholar
[33] Trasobares J, Vuillaume D, Théron D, Clément N 2016 Nat. Commun. 7 12850
Google Scholar
[34] Niu L L, Fu H Y, Suo Y Q, Liu R, Sun F, Wang S S, Zhang G P, Wang C K, Li Z L 2021 Phys. E 128 114542
Google Scholar
[35] Wu D, Huang L, Jia P Z, Cao X H, Fan Z Q, Zhou W X, Chen K Q 2021 Appl. Phys. Lett. 119 063503
Google Scholar
[36] Fan Z Q, Xie F, Jiang X W, Wei Z, Li S S 2016 Carbon 110 200
Google Scholar
[37] Kushmerick J 2009 Nature 462 994
Google Scholar
[38] Richter S, Mentovich E, Elnathan R 2018 Adv. Mater. 30 1706941
Google Scholar
[39] Liu R, Han Y, Sun F, Khatri G, Kwon J, Nickle C, Wang L, Wang C K, Thompson D, Li Z L, Nijhuis C A, del Barco E 2022 Adv. Mater. 34 2202135
Google Scholar
[40] Fan Z Q, Sun W Y, Jiang X W, Zhang Z H, Deng X Q, Tang G P, Xie H Q, Long M Q 2017 Carbon 113 18
Google Scholar
[41] Zhou Q, Yang Y, Ni J, Li Z, Zhang Z 2010 Nano Res. 3 423
Google Scholar
[42] Baghernejad M, Van Dyck C, Bergfield J, Levine D R, Gubicza A, Tovar J D, Calame M, Broekmann P, Hong W 2019 Chem. Eur. J. 25 15141
Google Scholar
[43] Büttiker M, Imry Y, Landauer R, Pinhas S 1985 Phys. Rev. B 31 6207
Google Scholar
[44] Smidstrup S, Markussen T, Vancraeyveld P, Wellendorff J, Schneider J, Gunst T, Verstichel B, Stradi D, Khomyakov P A, Vej-Hansen U G, Lee M E, Chill S T, Rasmussen F, Penazzi G, Corsetti F, Ojanperä A, Jensen K, Palsgaard M L N, Martinez U, Blom A, Brandbyge M, Stokbro K 2019 J. Phys. Condens. Matter 32 015901
Google Scholar
[45] Pan H, Tang L M, Chen K Q 2022 Phys. Rev. B 105 064401
Google Scholar

 下载:
下载:
计量
- 文章访问数: 6019
- PDF下载量: 64
- 被引次数: 0
