











-
Silicate cathode material Li2CoSiO4 has received wide attention due to high theoretical capacity. However, the high discharge makes the existing electrolyte unable to satisfy the requirements of its use, and the poor cyclic stability limits its further application and development. The high discharge and cycle stability of Li2CoSiO4 cathode material can be improved by doping corresponding elements. The effects of non-transition high-valent elements of Ga, Ge and As doping on structural, electrochemical and electronic properties of Li-ion battery cathode material Li2CoSiO4 are systematically studied by the first-principles calculations based on density functional theory within the generalized gradient approximation with Hubbard corrections (GGA + U). The calculation results show that the maximum expansion range of the unit cell volume of Li2CoSiO4 cathode material during lithium ion removal is 3.5%. However, the Ga, Ge and As doping reduce the variation range of unit cell volume during the delithiation of the system, which is beneficial to the improvement of the cycle stability of Li2CoSiO4 material. Furthermore, the Ga, Ge and As doping can reduce the theoretical average deintercalation voltages of extraction for the first Li+ in per formula unit; the theoretical average deintercalation voltages of the doping systems decrease by 1.65 V, 1.64 V and 1.64 V, respectively, compared with the deintercalation voltage of the undoped Li2CoSiO4 system. Meanwhile, except for the Ga doping, the Ge and As doping can also effectively reduce their theoretical average deintercalation voltagesin the secondary delithiation process. The density of states and magnetic moment show that Co2+ has a strong binding effect on the 3d orbital electrons, which makes it difficult for Co2+ in Li2CoSiO4 material to lose electrons for participating in the charge compensation in the process of Li+ removal. However, the Ga, Ge and As doping can effectively participate in the charge compensation of the system in the process of Li+ removal, which is the main reason for the decrease of the theoretical average deintercalation voltage of the system. In addition, the Ge doping reduces the band gap value of the Li2CoSiO4 from 3.7 eV to 2.49 eV, while the Ga doping and the As doping introduce the donor defects, and thus making the doping system exhibit metallic properties, which can improve the conductivity of the system to some extent.
[1] Larcher D, Tarascon J M 2015 Nat. Chem. 7 19
[2] Meng Y S, Dompablo M E A 2009 Energy Environ. Sci. 2 589
 Google Scholar
Google Scholar
[3] Ding Y F, Zhao Q Q, Yu Z L, Zhao Y Q, Liu B, He P B, Zhou H, Li K L, Yin S F, Cai M Q 2019 J. Mater. Chem. C 7 7433
Google Scholar
[4] Deng X Z, Zhao Q Q, Zhao Y Q, Cai M Q 2019 Curr. Appl. Phys. 19 279
Google Scholar
[5] Xu B, Qian D, Wang Z, Meng Y S 2012 Mater. Sci. Eng. R-Rep. 73 51
Google Scholar
[6] Zhao Y Q, Wang X, Liu B, Yu Z L, He P B, Wan Q, Cai M Q, Yu H L 2018 Org. Electron. 53 50
Google Scholar
[7] Zhao Y Q, Ma Q R, Liu B, Yu Z L, Yang J L, Cai M Q 2018 Nanoscale 10 8677
Google Scholar
[8] Dominko R, Bele M, Kokalj A, Gaberscek M, Jamnik J 2007 J. Power Sources 174 457
Google Scholar
[9] Sasaki H, Nemoto A, Moriya M, Miyahara M, Hokazono M, Katayama S, Akimoto Y, Nakajima A, Hirano S I 2015 Ceram. Int. 41 S680
Google Scholar
[10] Lyness C, Delobel B, Robert A A, Bruce P G 2007 Chem. Commun. 46 4890
[11] 嘉明珍 2017 博士学位论文(成都: 西南交通大学)
Jia M Z 2017 Ph. D. Dissertation (Chengdu: Southwest Jiaotong University) (in Chinese)
[12] Zhang Z F, Chen Z L, Zhang X H, Wu D Y, Li J 2018 Electrochim. Acta 264 166
Google Scholar
[13] Wu S Q, Zhu Z Z, Yang Y, Hou Z F 2009 Trans. Nonferrous Met. Soc. 19 182
Google Scholar
[14] Du H W, Zhang X H, Chen Z L, Wu D Y, Zhang Z F, Li J 2018 RSC Adv. 8 22813
[15] Kresse G, Joubert D 1999 Phys. Rev. B 59 1758
[16] Kresse G, Furthmüller J 1996 Comput. Mater. Sci. 6 15
Google Scholar
[17] Kresse G, Furthmüller J 1996 Phys. Rev. B 54 11169
Google Scholar
[18] Perdew J P, Burke K, Ernzerhof M 1996 Phys. Rev. Lett. 77 3865
Google Scholar
[19] Anisimov V I, Zaanen J, Andersen O K 1991 Phys. Rev. B 44 943
Google Scholar
[20] Zhou F, Cococcioni M, Marianetti C A, Morgan D, Ceder G 2004 Phys. Rev. B 70 235121
Google Scholar
[21] Robert A A, Lyness C, Ménétrier M, Bruce P G 2010 Chem. Mater. 22 1892
Google Scholar
[22] Zhou F, Cococcioni M, Kang K, Ceder G 2004 Electrochem. Commun. 6 1144
Google Scholar
[23] Graetz J, Hightower A, Ahu C C, Yazami R, Rez P, Fultz B 2002 J. Phys. Chem. B 106 1286
[24] Marianetti C A, Kotliar G, Ceder G 2004 Phys. Rev. Lett. 92 196405
Google Scholar
[25] Zhong G H, Li Y L, Yan P, Liu Z, Xie M H, Lin H Q 2010 J. Phys. Chem. C 114 3693
Google Scholar
[26] Li L, Zhu L, Xu L H, Cheng T M, Wang W, Li X, Sui Q T 2014 J. Mater. Chem. A 2 4251
Google Scholar
[27] Zhang P, Hu C H, Wu S Q, Zhu Z Z, Yang Y 2012 Phys. Chem. Chem. Phys. 14 7346
Google Scholar
[28] Chakrabarti S, Thakur A K, Biswas K 2017 Electrochim. Acta 236 288
Google Scholar
[29] Li Y S, Cheng X, Zhang Y 2013 Electrochim. Acta 112 670
Google Scholar
[30] Wu S Q, Zhang J H, Zhu Z Z, Yang Y 2007 Curr. Appl. Phys. 7 611
Google Scholar
[31] 嘉明珍, 王红艳, 陈元正, 马存良, 王辉 2015 物理学报 64 087101
Google Scholar
Jia M Z, Wang H Y, Chen Y Z, Ma C L, Wang H 2015 Acta Phys. Sin. 64 087101
Google Scholar
[32] Zhang P, Zheng Y, Wu S Q, Zhu Z Z, Yang Y 2014 Comput. Mater. Sci. 83 45
Google Scholar
[33] Huang Y L, Fan W B, Hou Y H, Guo K X, Ouyang Y F, Liu Z W 2017 J. Magn. Magn. Mater. 429 263
Google Scholar
[34] Boyd R J, Markus G E 1981 J. Chem. Phys. 75 5385
Google Scholar
[35] Pauling L 1960 The Nature of The Chemical Bond (London: Oxford University Press) p100
-

图 1 (a) Li2Co0.5R0.5SiO4 (R = Co, Ga, Ge, As)晶胞结构; (b)相应的超胞结构(绿色、粉色、橙色和蓝色四面体分别表示LiO4, CoO4, RO4和SiO4)
Figure 1. (a) Crystal cell structure of Li2Co0.5R0.5SiO4 (R = Co, Ga, Ge and As); (b) the corresponding supercell. Green, pink, orange and blue tetrahedron represent LiO4, CoO4, RO4 and SiO4, respectively.
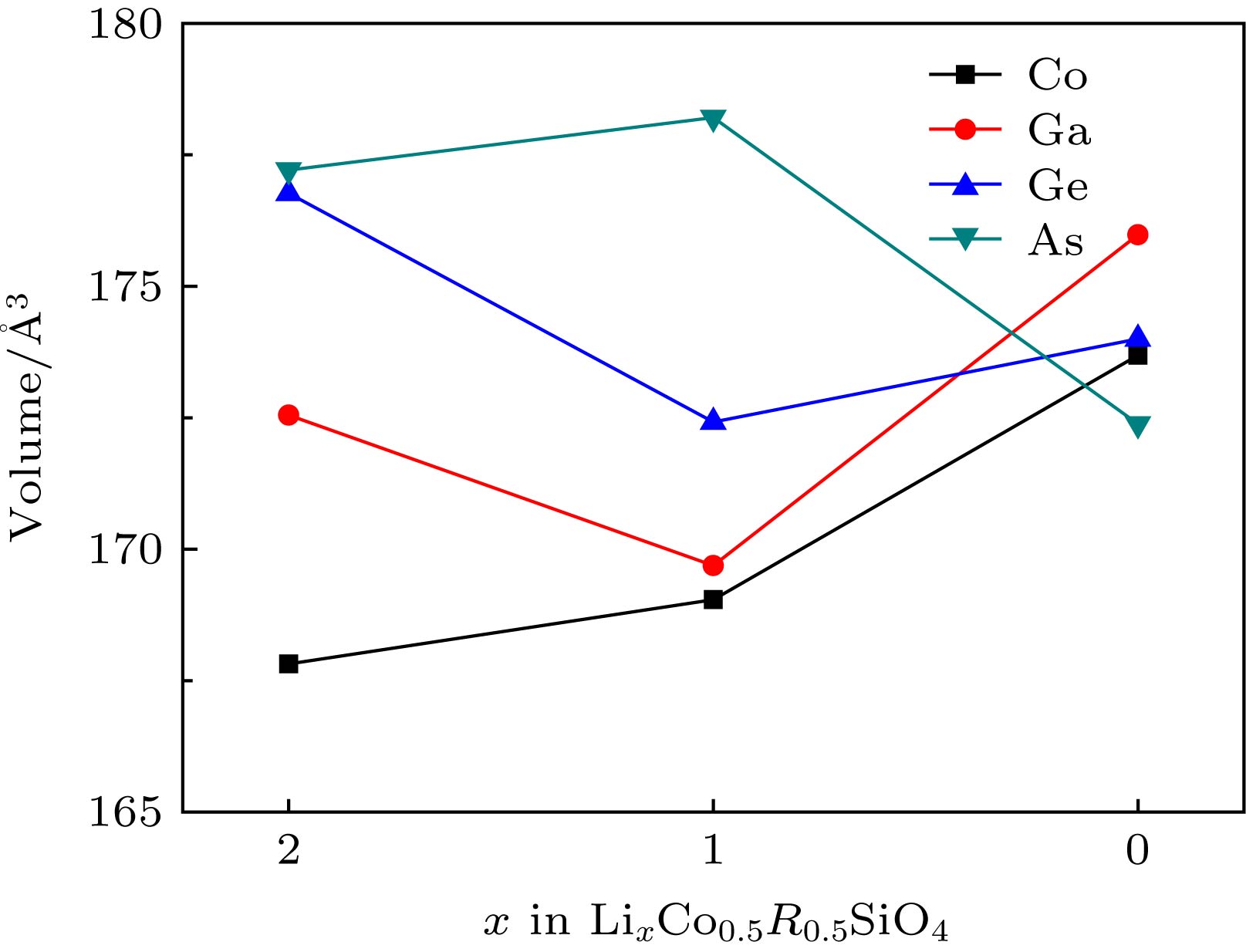
图 2 LixCo0.5R0.5SiO4 (R = Co, Ga, Ge, As; x = 0, 1, 2)脱锂过程中体积的变化
Figure 2. Corresponding unit cell volume of LixCo0.5R0.5SiO4 (R = Co, Ga, Ge and As) at during delithiation x (x = 0, 1, 2).
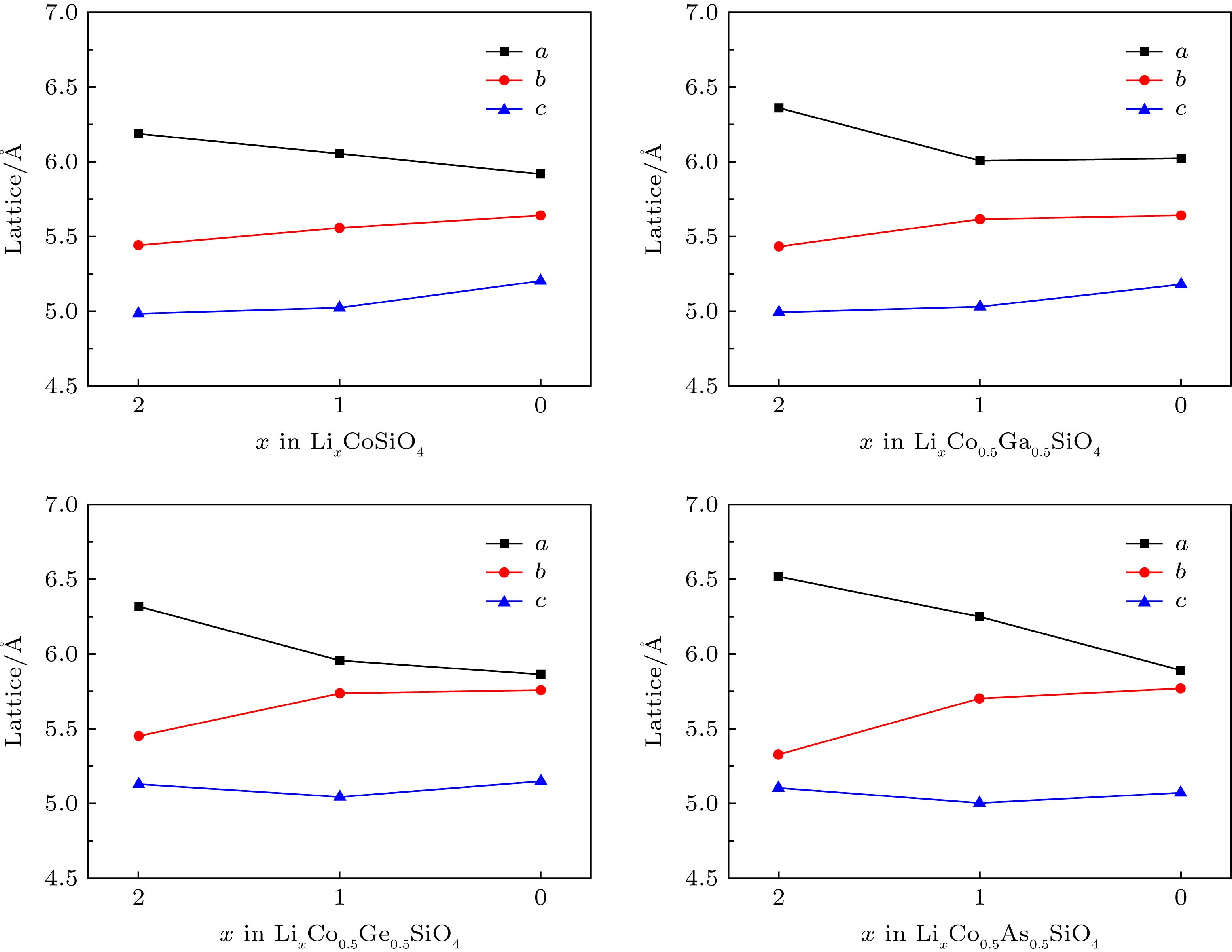
图 3 LixCo0.5R0.5SiO4 (R = Co, Ga, Ge, As; x = 0, 1, 2)脱锂过程中晶格常数a, b和c的变化
Figure 3. Variations of lattice parameters a, b and c of LixCo0.5R0.5SiO4 (R = Co, Ga, Ge, As) at during delithiation x (x = 0, 1, 2).

图 4 Li2Co0.5R0.5SiO4 (R = Co, Ga, Ge, As)体系的理论平均脱嵌电压
Figure 4. Average deintercalation voltages of Li2Co0.5R0.5SiO4 (R = Co, Ga, Ge and As).
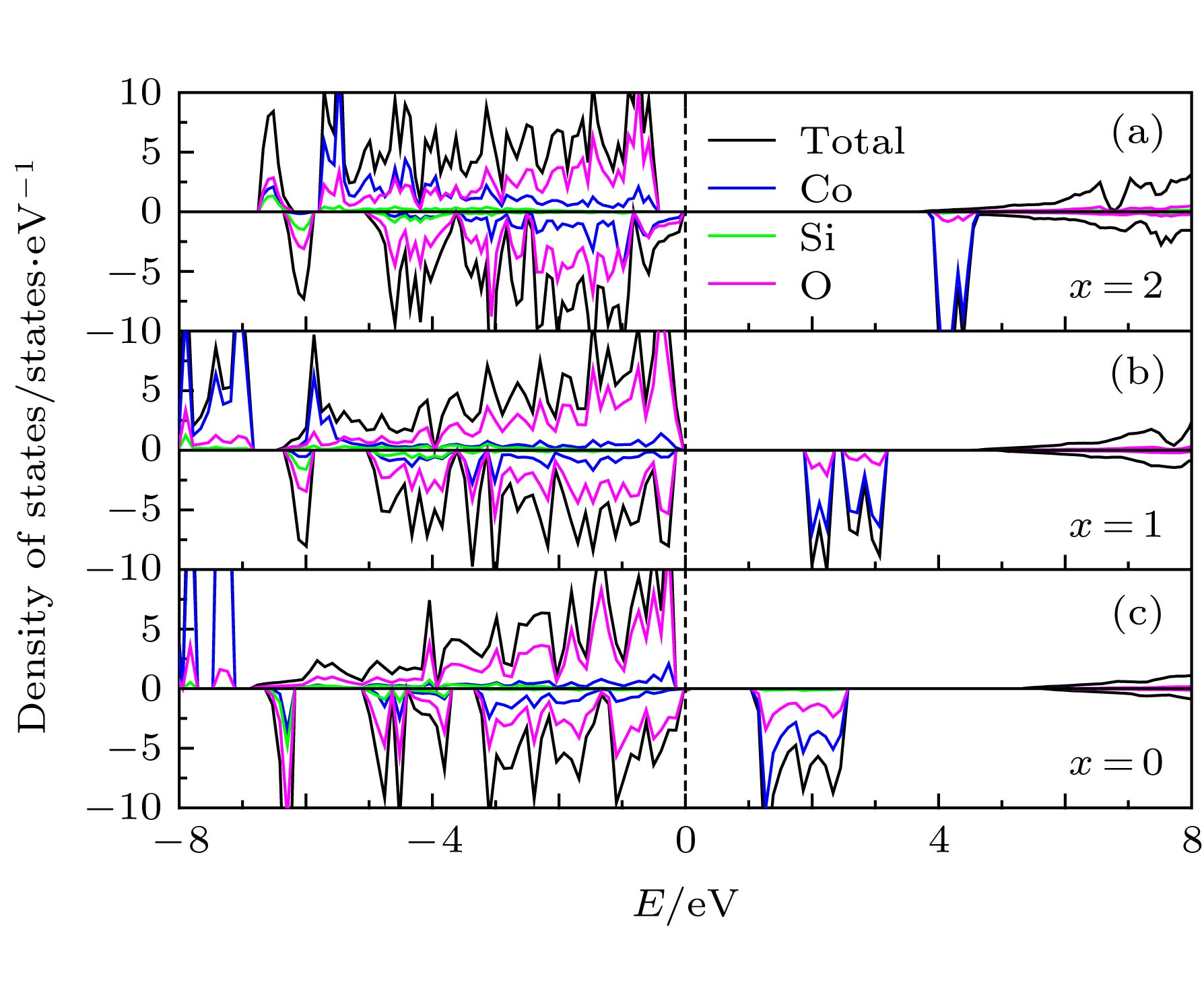
图 5 脱锂结构的态密度图 (a) Li2CoSiO4; (b) LiCoSiO4; (c) CoSiO4
Figure 5. Density of states of (a) Li2CoSiO4; (b) LiCoSiO4; (c) CoSiO4.
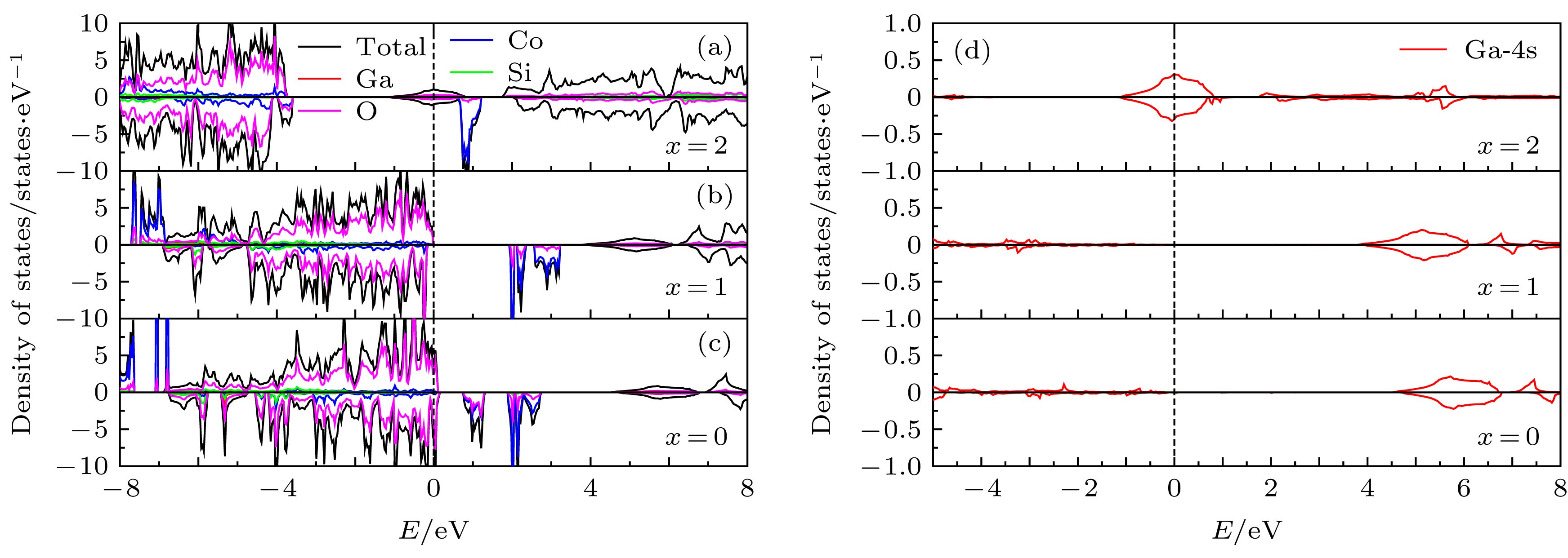
图 6 脱锂结构(a) Li2Co0.5Ga0.5SiO4, (b) LiCo0.5Ga0.5SiO4, (c) Co0.5Ga0.5SiO4的态密度图; (d)在脱锂过程中LixCo0.5Ga0.5SiO4(x = 0, 1, 2)中Ga的PDOS图
Figure 6. Density of states of (a) Li2Co0.5Ga0.5SiO4, (b) LiCo0.5Ga0.5SiO4, (c) Co0.5Ga0.5SiO4; (d) the PDOS of Ga in LixCo0.5Ga0.5SiO4 (x = 0, 1, 2) during delithiation.

图 7 脱锂结构(a) Li2Co0.5Ge0.5SiO4, (b) LiCo0.5Ge0.5SiO4, (c) Co0.5Ge0.5SiO4的态密度图; (d)表示在脱锂过程中LixCo0.5Ge0.5SiO4 (x = 0, 1, 2)中Ge的PDOS
Figure 7. Density of states of (a) Li2Co0.5Ge0.5SiO4, (b) LiCo0.5Ge0.5SiO4, (c) Co0.5Ge0.5SiO4; (d) the PDOS of Ge in LixCo0.5Ge0.5SiO4 (x = 0, 1, 2) during delithiation.
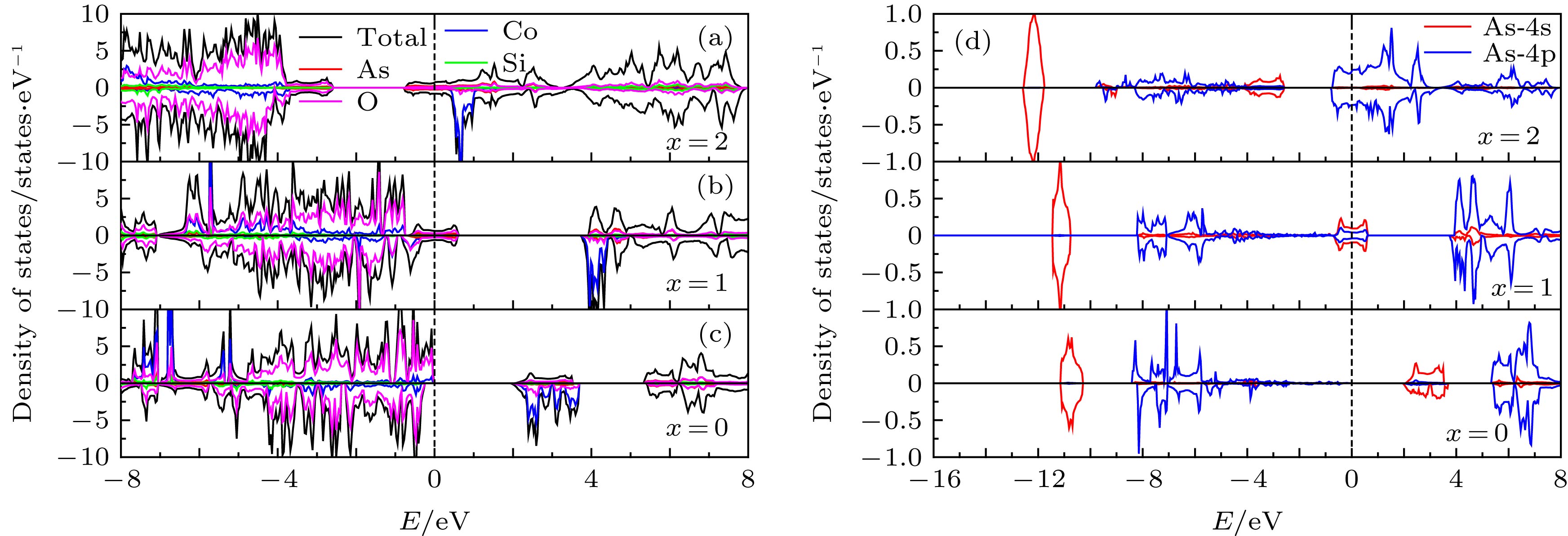
图 8 脱锂结构(a) Li2Co0.5As0.5SiO4, (b) LiCo0.5As0.5SiO4, (c) Co0.5As0.5SiO4的态密度图; (d)在脱锂过程中LixCo0.5As0.5SiO4 (x = 0, 1, 2)中As的PDOS
Figure 8. Density of states of (a) Li2Co0.5As0.5SiO4, (b) LiCo0.5As0.5SiO4, (c) Co0.5As0.5SiO4; (d) the PDOS of As in LixCo0.5As0.5SiO4 (x = 0, 1, 2) during delithiation.
表 1 Co离子的磁矩和氧化态
Table 1. Magnetic moment and oxidation state of Co ion.
结构 磁矩/μB 氧化态 Li2CoSiO4 2.79 +2 (4s03d7) LiCoSiO4 3.18 +3 (4s03d6) CoSiO4 3.34 +3 (4s03d6)  DownLoad: CSV
DownLoad: CSV
表 2 Co离子的磁矩和氧化态
Table 2. Magnetic moment and oxidation state of Co ion.
结构 磁矩/μB 氧化态 Li2Co0.5Ga0.5SiO4 2.79 +2 (4s03d7) LiCo0.5Ga0.5SiO4 3.18 +3 (4s03d6) Co0.5Ga0.5SiO4 3.26 +3 (4s03d6)
DownLoad: CSV
表 3 Co离子的磁矩和氧化态
Table 3. Magnetic moment and oxidation state of Co ion.
结构 磁矩/μB 氧化态 Li2Co0.5Ge0.5SiO4 2.79 +2 (4s03d7) LiCo0.5Ge0.5SiO4 2.79 +2 (4s03d7) Co0.5Ge0.5SiO4 3.35 +3 (4s03d6)
DownLoad: CSV
表 4 Co离子的磁矩和氧化态
Table 4. Magnetic moment and oxidation state of Co ion.
结构 磁矩/μB 氧化态 Li2Co0.5As0.5SiO4 2.79 +2 (4s03d7) LiCo0.5As0.5SiO4 2.79 +2 (4s03d7) Co0.5As0.5SiO4 3.26 +3 (4s03d6)
DownLoad: CSV
-
[1] Larcher D, Tarascon J M 2015 Nat. Chem. 7 19
[2] Meng Y S, Dompablo M E A 2009 Energy Environ. Sci. 2 589
Google Scholar
[3] Ding Y F, Zhao Q Q, Yu Z L, Zhao Y Q, Liu B, He P B, Zhou H, Li K L, Yin S F, Cai M Q 2019 J. Mater. Chem. C 7 7433
Google Scholar
[4] Deng X Z, Zhao Q Q, Zhao Y Q, Cai M Q 2019 Curr. Appl. Phys. 19 279
Google Scholar
[5] Xu B, Qian D, Wang Z, Meng Y S 2012 Mater. Sci. Eng. R-Rep. 73 51
Google Scholar
[6] Zhao Y Q, Wang X, Liu B, Yu Z L, He P B, Wan Q, Cai M Q, Yu H L 2018 Org. Electron. 53 50
Google Scholar
[7] Zhao Y Q, Ma Q R, Liu B, Yu Z L, Yang J L, Cai M Q 2018 Nanoscale 10 8677
Google Scholar
[8] Dominko R, Bele M, Kokalj A, Gaberscek M, Jamnik J 2007 J. Power Sources 174 457
Google Scholar
[9] Sasaki H, Nemoto A, Moriya M, Miyahara M, Hokazono M, Katayama S, Akimoto Y, Nakajima A, Hirano S I 2015 Ceram. Int. 41 S680
Google Scholar
[10] Lyness C, Delobel B, Robert A A, Bruce P G 2007 Chem. Commun. 46 4890
[11] 嘉明珍 2017 博士学位论文(成都: 西南交通大学)
Jia M Z 2017 Ph. D. Dissertation (Chengdu: Southwest Jiaotong University) (in Chinese)
[12] Zhang Z F, Chen Z L, Zhang X H, Wu D Y, Li J 2018 Electrochim. Acta 264 166
Google Scholar
[13] Wu S Q, Zhu Z Z, Yang Y, Hou Z F 2009 Trans. Nonferrous Met. Soc. 19 182
Google Scholar
[14] Du H W, Zhang X H, Chen Z L, Wu D Y, Zhang Z F, Li J 2018 RSC Adv. 8 22813
[15] Kresse G, Joubert D 1999 Phys. Rev. B 59 1758
[16] Kresse G, Furthmüller J 1996 Comput. Mater. Sci. 6 15
Google Scholar
[17] Kresse G, Furthmüller J 1996 Phys. Rev. B 54 11169
Google Scholar
[18] Perdew J P, Burke K, Ernzerhof M 1996 Phys. Rev. Lett. 77 3865
Google Scholar
[19] Anisimov V I, Zaanen J, Andersen O K 1991 Phys. Rev. B 44 943
Google Scholar
[20] Zhou F, Cococcioni M, Marianetti C A, Morgan D, Ceder G 2004 Phys. Rev. B 70 235121
Google Scholar
[21] Robert A A, Lyness C, Ménétrier M, Bruce P G 2010 Chem. Mater. 22 1892
Google Scholar
[22] Zhou F, Cococcioni M, Kang K, Ceder G 2004 Electrochem. Commun. 6 1144
Google Scholar
[23] Graetz J, Hightower A, Ahu C C, Yazami R, Rez P, Fultz B 2002 J. Phys. Chem. B 106 1286
[24] Marianetti C A, Kotliar G, Ceder G 2004 Phys. Rev. Lett. 92 196405
Google Scholar
[25] Zhong G H, Li Y L, Yan P, Liu Z, Xie M H, Lin H Q 2010 J. Phys. Chem. C 114 3693
Google Scholar
[26] Li L, Zhu L, Xu L H, Cheng T M, Wang W, Li X, Sui Q T 2014 J. Mater. Chem. A 2 4251
Google Scholar
[27] Zhang P, Hu C H, Wu S Q, Zhu Z Z, Yang Y 2012 Phys. Chem. Chem. Phys. 14 7346
Google Scholar
[28] Chakrabarti S, Thakur A K, Biswas K 2017 Electrochim. Acta 236 288
Google Scholar
[29] Li Y S, Cheng X, Zhang Y 2013 Electrochim. Acta 112 670
Google Scholar
[30] Wu S Q, Zhang J H, Zhu Z Z, Yang Y 2007 Curr. Appl. Phys. 7 611
Google Scholar
[31] 嘉明珍, 王红艳, 陈元正, 马存良, 王辉 2015 物理学报 64 087101
Google Scholar
Jia M Z, Wang H Y, Chen Y Z, Ma C L, Wang H 2015 Acta Phys. Sin. 64 087101
Google Scholar
[32] Zhang P, Zheng Y, Wu S Q, Zhu Z Z, Yang Y 2014 Comput. Mater. Sci. 83 45
Google Scholar
[33] Huang Y L, Fan W B, Hou Y H, Guo K X, Ouyang Y F, Liu Z W 2017 J. Magn. Magn. Mater. 429 263
Google Scholar
[34] Boyd R J, Markus G E 1981 J. Chem. Phys. 75 5385
Google Scholar
[35] Pauling L 1960 The Nature of The Chemical Bond (London: Oxford University Press) p100

 DownLoad:
DownLoad:
Catalog
Metrics
- Abstract views: 14235
- PDF Downloads: 131
- Cited By: 0
