










-
基于Molpro 2012程序包, 应用包含Davidson修正的多参考组态相互作用方法, 使用AVX Z和AVX dZ (X = T, Q, 5, 6)基组进行单点能从头算, 然后采用Aguado-Paniagua函数进行拟合, 得到了SiH+(X1Σ+)离子在不同基组、不同方法和是否考虑自旋-轨道耦合(SOC)情况下的解析势能函数(APEFs). 以APEFs为基础, 计算了SiH+(X1Σ+)离子的解离能De, 平衡键长Re, 振动频率ωe, 光谱常数Be, αe和ωeχe, 同时讨论了SOC对该体系的影响. 本文的计算结果与其他理论计算符合得较好, 与实验数值也基本吻合. 基于SOC-AV6dZ方法下的APEF, 通过求解径向薛定谔方程, 给出了SiH+(X1Σ+)离子的前23个振动能级(j = 0), 并详细列出了每1个振动能级及其相应的经典拐点, 每个振动态的转动常数和6个离心畸变常数, 且提供了振动能级图. 该工作对于实验和后续的理论工作有参考和指导作用.
-
关键词:
- SiH+(X1Σ+)离子 /
- 势能曲线 /
- 光谱常数 /
- 自旋-轨道耦合
The analytical potential energy function (APEF) of SiH+(X1Σ+) is fitted by Aguado-Paniagua function with 112 ab initio energy points, which are calculated by Molpro 2012 Package with the multi-reference configuration interaction including the Davidson correction method using AVX Z and AVX dZ (X = Q, 5, 6) basis sets. Moreover, the calculated ab initio energy points are subsequently extrapolated to complete basis set (CBS) limit to avoid the basis set superposition error. All the fitting parameters of APEFs for AV6Z, CBS(Q, 5), AV6dZ, CBS(Qd, 5d), SA-AV6dZ and SOC-AV6dZ methods are gathered. The potential energy curves (PEC) and the corresponding ab initio points are also shown. As can be seen, the PECs show excellent agreement with the ab initio points and a smooth behavior both in short range and long range, which ensures the high quality of fitting process for the current APEFs. Based on these APEFs of different basis sets and methods including AVQZ, AV5Z, AV6Z, CBS(Q, 5), AVQdZ, AV5dZ, AV6dZ and CBS(Qd, 5d), the spectral constants De, Re, ωe, Be, αe and ωeχe are obtained. In addition, the effects of spin-orbit coupling interaction (SOC) on the system are also investigated. By comparing the spectral constants of SA-AV6dZ with the ones of SOC-AV6dZ, it is found that the effect of SOC on SiH+(X1Σ+) is small and can be ignored. We also compare the spectral constants in this work with the experimental values and other theoretical results. The results of this work accord well with the corresponding experimental and other theoretical results. It is worth noting that the deviation of dissociation energy between the theoretical calculations and experimental values is relatively large. Based on this conclusion, we suggest that the spectral constants including the dissociation energy for SiH+(X1Σ+) should be remeasured. Based on the APEF of SOC-AV6dZ which should be more accurate than others in theory, the top 23 vibrational states (j = 0) of SiH+(X1Σ+) are calculated first by solving the radial Schrödinger equation. All the vibrational energy levels, classical turning points, rotation constants and six centrifugal distortion constants are also provided. The results of this work can provide significant references for the experimental and other theoretical work. -
Keywords:
- SiH+(X1Σ+) ion /
- potential energy curve /
- spectral constant /
- spin-orbit coupling interaction
[1] Grevesse N, Sauval A J 1970 Astron. Astrophys. 9 232
[2] Douglas A E, Lutz B L 1970 Can. J. Phys. 48 247
 Google Scholar
Google Scholar
[3] Grevesse N, Sauval A J 1971a J. Quant. Spectrosc. Radiat. Transf. 11 65
Google Scholar
[4] Almeida A A, Sing P D 1978 Astrophys. Space Sci. 56 415
Google Scholar
[5] Gao W, Wang B B, Hu X J, Chai S, Han Y C, Greenwood J B 2017 Phys. Rev. A 96 013426
Google Scholar
[6] Wang B B, Han Y C, Gao W, Cong S L 2017 Phys. Chem. Chem. Phys. 19 22926
Google Scholar
[7] Moore P L, Browne J C, Matsen F A 1965 J. Chem. Phys. 43 903
Google Scholar
[8] Cosby P C, Helm H, Moseley J T 1980 Astrophys. J. 235 52
Google Scholar
[9] Barinovs G, Hemert M C V 2006 Astrophys. J. 636 923
Google Scholar
[10] Ram R S, Engleman R, Bernath P F 1998 J. Mol. Spectrosc. 190 341
Google Scholar
[11] Singh P D, Vanlandingham F G 1978 Astron. Astrophys. 66 87
Google Scholar
[12] Carlson T A, Copley J, Duric N, Elander N, Erman P, Larsson M, Lyyra M 1980 Astron. Astrophys. 83 238
[13] Hishikawa A, Karawajczyk A 1993 J. Mol. Spectrosc. 158 479
Google Scholar
[14] Davies P B, Martineau P M 1988 J. Chem. Phys. 88 485
Google Scholar
[15] Mosnier J P, Kennedy E T, Kampen P V, Cubaynes D, Guilbaud S, Sisourat N, Puglisi A, Carniato S, Bizau J M 2016 Phys. Rev. A 93 061401
Google Scholar
[16] Hirst D M 1986 Chem. Phys. Lett. 128 504
Google Scholar
[17] Langhoff S R, Davidson E R 1974 Int. J. Quantum Che. 8 61
Google Scholar
[18] Matos J M O, Kello V, Roos B O, Sadlej A J 1988 J. Chem. Phys. 89 423
Google Scholar
[19] Sannigrahi A B, Buenker R J, Hirsch G, Gu J P 1995 Chem. Phys. Lett. 237 204
Google Scholar
[20] Werner H J, Knowles P J, Lindh R, Manby F R, Schutz M, et al. Molpro, A Package of ab initio Programs (Version 2015.1) http://www.molpro.net [2021-03-08]
[21] Zhang Y G, Dou G, Cui J, Yu Y 2018 J. Mol. Struct. 1165 318
Google Scholar
[22] Neese F 2011 Wiley Interdisci. Rev. Comput. Mol. Sci. 2 73
[23] Biglari Z, Shayesteh A, Ershadifar S 2018 J. Quant. Spectrosc. Radiat. Transf. 221 80
Google Scholar
[24] Werner H J, Knowles P J, Lindh R, Manby F R, Schutz M, et al. Molpro, A Package of ab initio Programs (Version 2012.1) http://www.molpro.net [2021-03-08]
[25] Aguado A, Paniagua M 1992 J. Chem. Phys. 96 1265
Google Scholar
[26] Aguado A, Tablero C, Paniagua M 1998 Comput. Phys. Commun. 108 259
Google Scholar
[27] Varandas A J C 2007 J. Chem. Phys. 126 244105
Google Scholar
[28] Varandas A J C 2000 J. Chem. Phys. 113 8880
Google Scholar
[29] Jansen H B, Ross P 1969 Chem. Phys. Lett. 3 140
Google Scholar
[30] Liu B, McLean A D 1973 J. Chem. Phys. 59 4557
Google Scholar
[31] Karton A, Martin J M L 2006 Theor. Chem. ACC. 115 330
Google Scholar
[32] Yang C L, Huang Y J, Zhang X, Han K L 2003 J. Mol. Struc. Theochem. 625 289
Google Scholar
[33] Yang C L, Zhang X, Han K L 2004 J. Mol. Struc. Theochem. 676 209
Google Scholar
[34] Huber K P, Herzberg G 1979 Molecular Spectra and Molecular Structure (Vol. IV) (New York: Springer) p600
[35] Roy R J L 2017 J. Quant. Spectrosc. Radiat. Transf. 186 167
Google Scholar
-
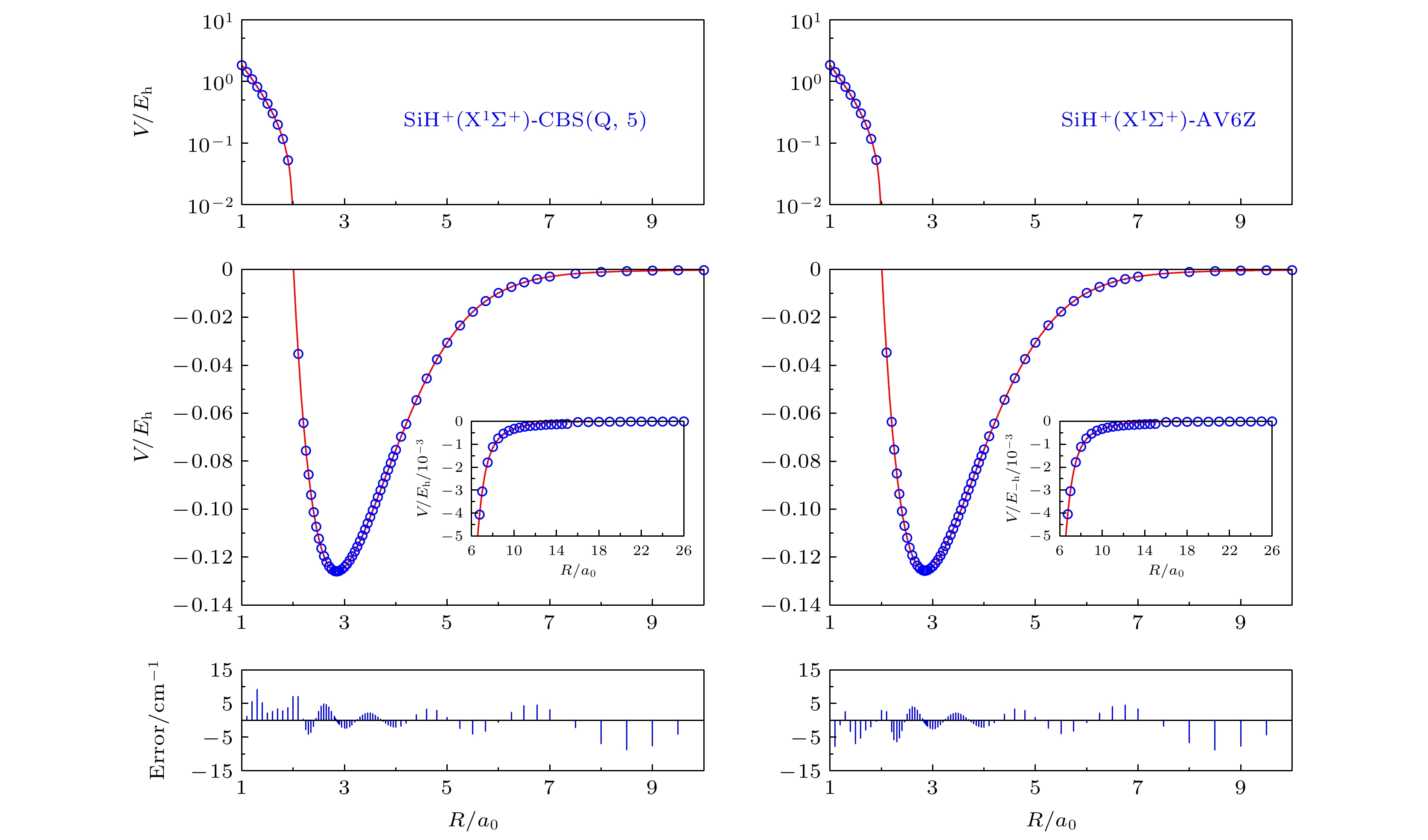
图 1 SiH+(X1Σ+)在CBS(Q, 5)和AV6Z基组下的势能曲线和从头算能量点
Fig. 1. Potential energy curves and ab initio points at CBS(Q, 5) and AV6Z results.
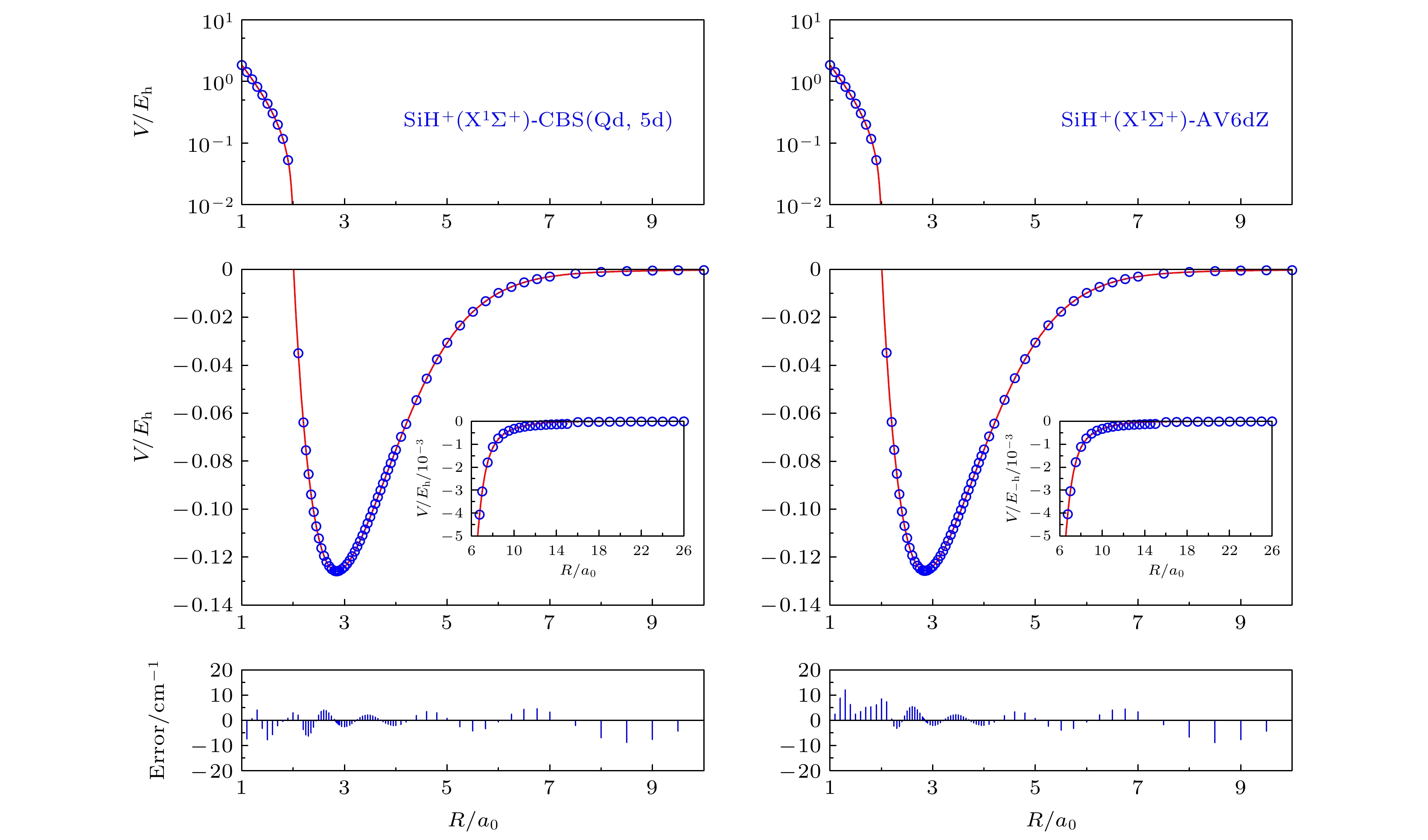
图 2 SiH+(X1Σ+)应用CBS(Qd, 5d)和AV6dZ基组的势能曲线和从头算能量点
Fig. 2. Potential energy curves and ab initio points at CBS(Qd, 5d) and AV6dZ results.
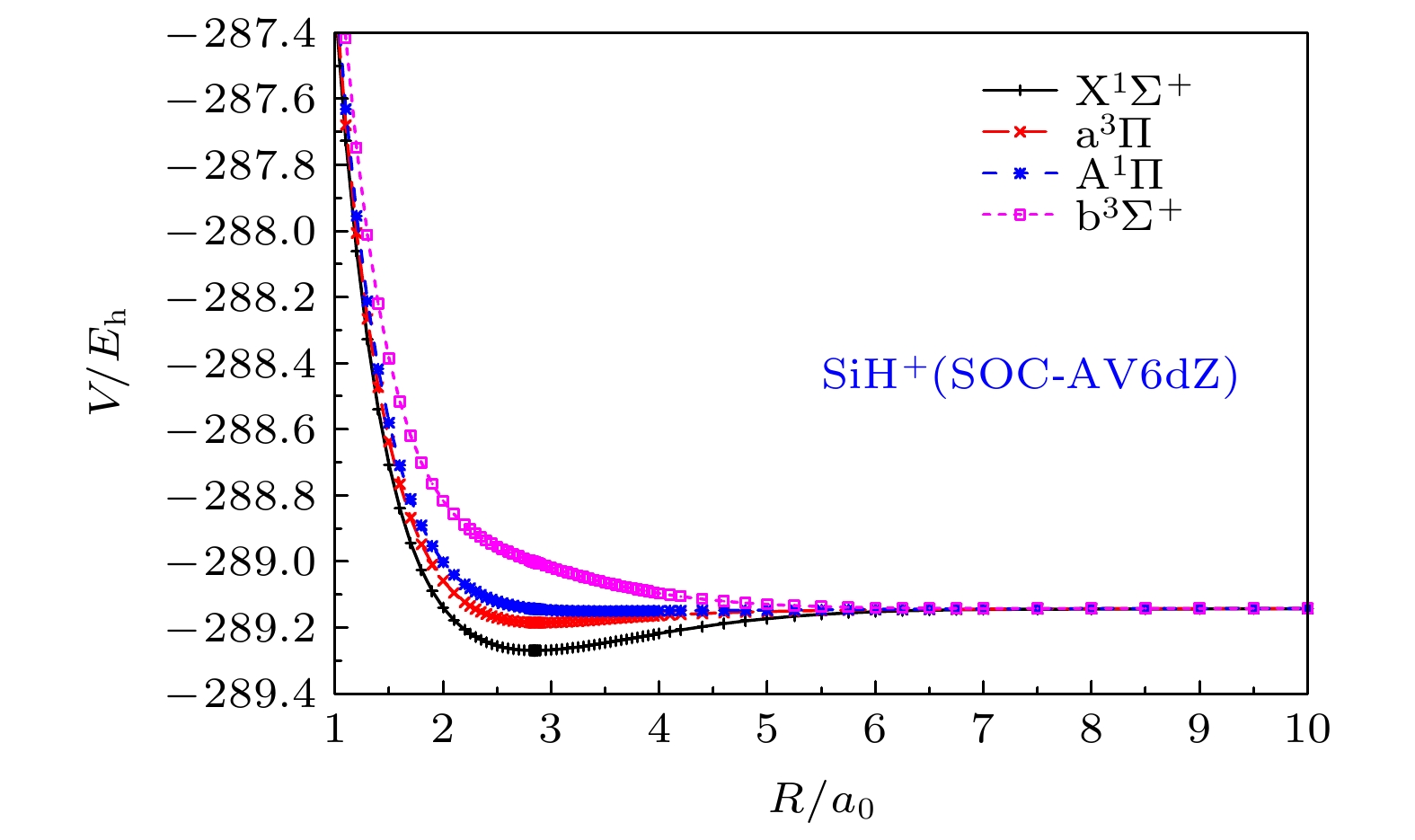
图 3 SiH+离子的
${{\rm{X}}^1}{\Sigma ^ + }$ ,${{\rm{A}}^1}\Pi$ ,${\rm{b}}{}^3{\Sigma ^ + }$ 和${{\rm{a}}^3}\Pi$ 态在SOC-AV6dZ基组下的从头算能量点Fig. 3. The ab initio points of
${{\rm{X}}^1}{\Sigma ^ + }$ ,${{\rm{A}}^1}\Pi$ ,${\rm{b}}{}^3{\Sigma ^ + }$ and${{\rm{a}}^3}\Pi$ states for SiH+ cation at SOC-AV6dZ results.
图 4 SiH+(X1Σ+)在SA-AV6dZ和SOC-AV6dZ基组下的从头算能量点与拟合势能曲线
Fig. 4. Potential energy curves and ab initio points at SA-AV6dZ and SOC-AV6dZ results.
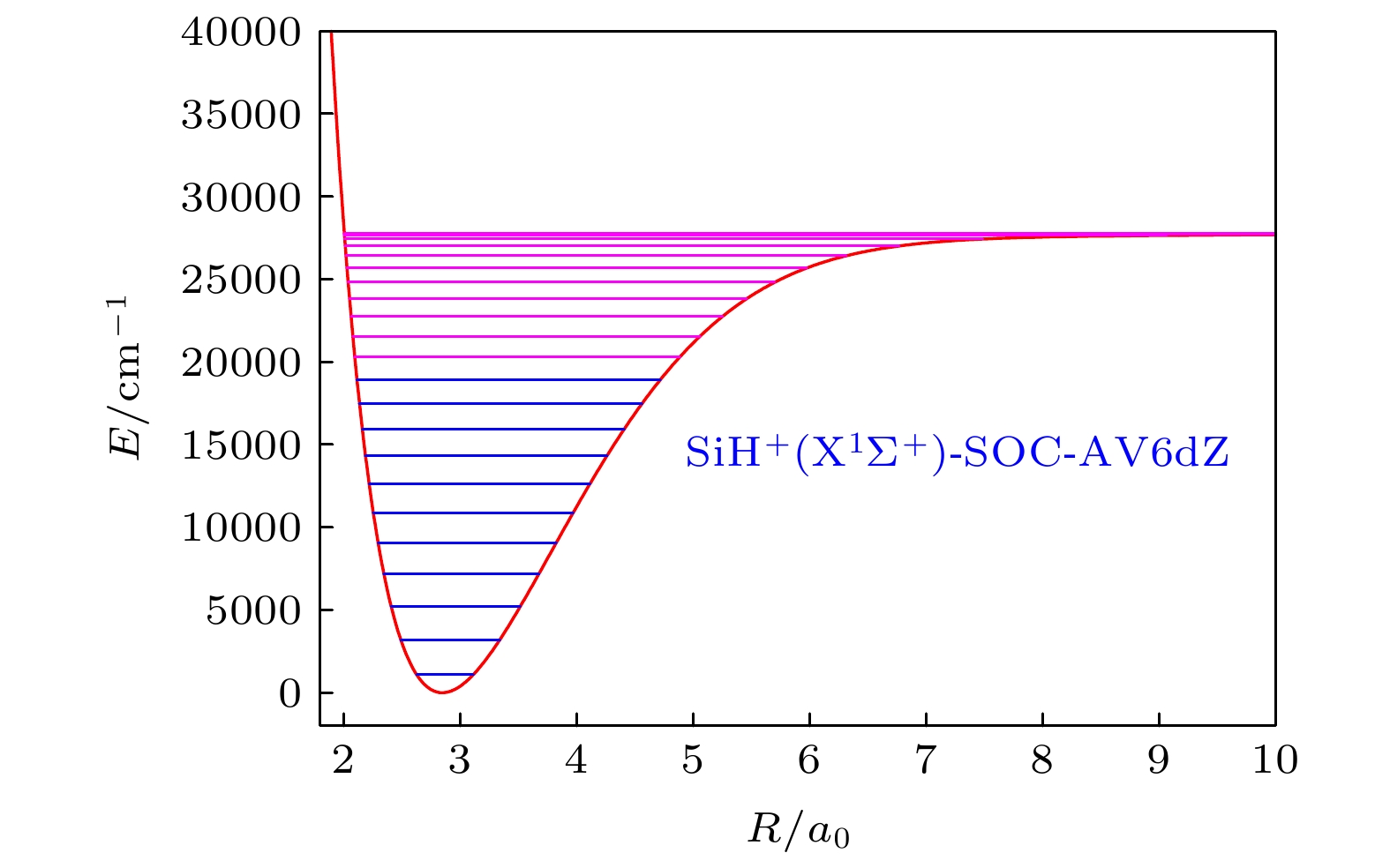
图 5 SiH+(X1Σ+)离子在SOC-AV6 dZ方法下, j = 0时的前23个振动能级
Fig. 5. Top 23 vibrational energy levels of SiH+(X1Σ+) when j = 0 at SOC-AV6 dZ result.
表 1 SiH+(X1Σ+) APEFs的拟合参数
Table 1. Parameters of APEFs for SiH+(X1Σ+).
AV6Z CBS(Q, 5) AV6dZ CBS(Qd, 5d) SA-AV6dZ SOC-AV6dZ a0 0.50876826×101 0.50552062×101 0.50858283×101 0.50691706×101 0.50773275×101 0.50772768×101 a1 –0.13804619×100 –0.14563907×100 –0.13801387×100 –0.14215220×100 –0.13720269×100 –0.15322183×100 a2 –0.13563746×101 –0.14531223×101 –0.13570613×101 –0.14215828×101 –0.16375066×101 0.30937784×100 a3 –0.51802913×102 –0.49687570×102 –0.51738027×102 –0.50317640×102 –0.42586263×102 –0.92938545×102 a4 0.62749955×103 0.59645776×103 0.62624067×103 0.60422890×103 0.45856480×103 0.11152320×104 a5 –0.48068238×104 –0.45486708×104 –0.47942732×104 –0.45972031×104 –0.30344319×104 –0.81653091×104 a6 0.26148147×105 0.24858810×105 0.26073093×105 0.24994077×105 0.14706913×105 0.40426343×105 a7 –0.98935973×105 –0.94946694×105 –0.98651660×105 –0.94886015×105 –0.51962969×105 –0.13681997×106 a8 0.24960293×106 0.24200403×106 0.24891691×106 0.24047472×106 0.12676883×106 0.31028426×106 a9 –0.39722347×106 –0.38879401×106 –0.39620641×106 –0.38442238×106 –0.19927330×106 –0.44979467×106 a10 0.35937312×106 0.35468541×106 0.35853489×106 0.34921672×106 0.18029764×106 0.37617755×106 a11 –0.14069370×106 –0.13986829×106 –0.14040272×106 –0.13721966×106 –0.71149416×106 –0.13802294×106 β1 0.6890 0.6800 0.6890 0.6840 0.6870 0.6870 β2 0.7470 0.7470 0.7470 0.7470 0.7470 0.7470 ∆Ermsd/
(kcal·mol–1)1.60176420×10–2 1.61755859×10–2 1.60042370×10–2 1.627559848×10–2 9.45767662×10–3 1.11170443×10–2  下载: 导出CSV
下载: 导出CSV
表 2 SiH+(X1Σ+)的平衡键长Re, 解离能De, 振动频率ωe, 光谱常数ωeχe, αe和βe
Table 2. Spectroscopic constants compared with the experimental values and other theoretical results for SiH+(X1Σ+).
基组 De(Eh) Re(a0) ωe/cm–1 βe/cm–1 αe/cm–1 ωeχe/cm–1 AVQZ 0.124640 2.851925 2153.245 7.607440 0.219327 42.373 AV5Z 0.125388 2.848589 2155.792 7.625272 0.218960 42.220 AV6Z 0.125584 2.848001 2156.686 7.628410 0.218833 42.189 CBS(Q, 5) 0.125856 2.847053 2157.189 7.633502 0.218622 42.117 AVQdZ 0.125067 2.848877 2156.187 7.623729 0.219427 42.343 AV5dZ 0.125461 2.848067 2156.737 7.628067 0.219011 42.232 AV6dZ 0.125622 2.847728 2157.046 7.629881 0.218849 42.190 CBS(Qd, 5d) 0.125801 2.847782 2156.602 7.629593 0.218534 42.112 SA-AV6dZ 0.127264 2.848531 2163.448 7.625581 0.216725 41.893 SOC-AV6dZ 0.126533 2.848382 2164.033 7.626378 0.217885 42.158 Expe[12,34] 0.123203 2.842338 2157.17 7.6603 0.2096 34.24 Theory[16] 0.118665 2.834590 2155.4 7.6786 0.2082 38.8 Theory[18] 0.123982 2.844039 2172.0 — — — Theory[21] 0.125317 2.834590 2177.9 7.6984 — 36.7 Theory[23] 0.124980 2.842149 2154.3 7.6609 0.2032 35.0
下载: 导出CSV
表 3 SiH+(X1Σ+)离子在SOC-AV6dZ方法下, j = 0时的前23个振动能级G(v)、经典拐点和惯性转动常数Bv
Table 3. Vibrational levels G(v), classical turn point androtational constant Bv for SiH+(X1Σ+) when j = 0 at SOC-AV6dZ result.
v G(v)/ cm–1 Rmin(a0) Rmax(a0) Bv/ cm–1 0 1074.467 2.62981 3.11141 7.530487 1 3171.224 2.49347 3.33862 7.336827 2 5200.543 2.40984 3.51594 7.142529 3 7162.231 2.34756 3.67498 6.947281 4 9055.742 2.29761 3.82512 6.750433 5 10880.124 2.25595 3.97091 6.551101 6 12634.005 2.22031 3.91505 6.348250 7 14315.600 2.18934 4.26009 6.140735 8 15922.726 2.16213 4.40742 5.927312 9 17452.802 2.13803 4.55893 5.706603 10 18902.835 2.11661 4.71650 5.477032 11 20269.364 2.09752 4.88227 5.236702 12 21548.363 2.08051 5.05888 4.983201 13 22735.066 2.06540 5.24986 4.713296 14 23823.694 2.05206 5.46016 4.422419 15 24807.010 2.04041 5.69731 4.103776 16 25675.599 2.03041 5.97379 3.746622 17 26416.625 2.02208 6.31278 3.332604 18 27011.579 2.01553 6.76545 2.826988 19 27432.546 2.01096 7.48310 2.158867 20 27650.956 2.00861 9.05975 1.323582 21 27734.462 2.00771 11.47553 0.838944 22 27769.256 2.00734 16.60576 0.360244
下载: 导出CSV
表 4 SiH+(X1Σ+)离子在SOC-AV6dZ方法下, j = 0时的前23个振动能级的6个离心畸变常数Dv, Hv, Lv, Mv, Nv和Ov
Table 4. Six centrifugal distortion constants Dv, Hv, Lv, Mv, Nv和Ov for the top 23 vibrational states of SiH+(X1Σ+) when j = 0 at SOC-AV6dZ result.
v Dv (× 10–4) Hv (× 10–8) Lv Mv Nv Ov 0 –3.7712102 1.4824556 –9.1045 × 10–13 4.1069 × 10–17 –2.4011 × 10–21 6.9631 × 10–26 1 –3.7362090 1.4243863 –8.8032 × 10–13 3.5297 × 10–17 –4.1863 × 10–21 1.8363 × 10–25 2 –3.7039865 1.3618539 –8.9866 × 10–13 2.9262 × 10–17 –5.1444 × 10–21 1.9171 × 10–25 3 –3.6778687 1.2884893 –9.4914 × 10–13 2.3397 × 10–17 –5.8593 × 10–21 9.6796 × 10–26 4 –3.6608093 1.2011917 –1.0216 × 10–12 1.6648 × 10–17 –6.8904 × 10–21 –1.1678 × 10–25 5 –3.6553038 1.0982022 –1.1140 × 10–12 6.8522 × 10–18 –8.7371 × 10–21 –4.8697 × 10–25 6 –3.6635199 0.9774540 –1.2320 × 10–12 –9.1302 × 10–18 –1.2003 × 10–20 –1.1229 × 10–24 7 –3.6875661 0.8351772 –1.3901 × 10–12 –3.5900 × 10–17 –1.7702 × 10–20 –2.2781 × 10–24 8 –3.7298613 0.6645057 –1.6143 × 10–12 –8.0925 × 10–17 –2.7799 × 10–20 –4.5064 × 10–24 9 –3.7936180 0.4536483 –1.9481 × 10–12 –1.5748 × 10–16 –4.6321 × 10–20 –9.0505 × 10–24 10 –3.8835055 1.8293995 –2.4663 × 10–12 –2.9088 × 10–16 –8.1928 × 10–20 –1.8869 × 10–23 11 –4.0066389 –0.1804552 –3.3026 × 10–12 –5.3251 × 10–16 –1.5445 × 10–19 –4.1577 × 10–23 12 –4.1741837 –0.6927552 –4.7113 × 10–12 –9.9426 × 10–16 –3.1311 × 10–19 –9.8710 × 10–23 13 –4.4041727 –1.4546951 –7.2135 × 10–12 –1.9417 × 10–15 –6.9300 × 10–19 –2.5871 × 10–22 14 –4.7268845 –2.6591579 –1.1978 × 10–11 –4.0783 × 10–15 –1.7160 × 10–18 –7.7417 × 10–22 15 –5.1961278 –4.7106022 –2.1959 × 10–11 –9.5527 × 10–15 –4.9431 × 10–18 –2.7809 × 10–21 16 –5.9157772 –8.5688303 –4.5904 × 10–11 –2.6336 × 10–14 –1.7665 × 10–17 –1.2991 × 10–20 17 –7.1117968 –16.938203 –1.1615 × 10–10 –9.3420 × 10–14 –8.7454 × 10–17 –9.0301 × 10–20 18 –9.3643712 –39.467832 –3.9578 × 10–10 –4.9264 × 10–13 –7.1396 × 10–16 –1.1444 × 10–18 19 –14.323856 –114.68916 –1.7949 × 10–9 –3.4005 × 10–12 –7.1696 × 10–15 –1.6154 × 10–17 20 –18.399913 –47.324191 1.5501 × 10–9 –7.5768 × 10–13 –3.7548 × 10–14 –1.9274 × 10–17 21 –14.637253 –291.40012 –2.0665 × 10–8 –1.4452 × 10–10 –1.3084 × 10–12 –1.3806 × 10–14 22 –57.213156 –20756.133 –1.6655 × 10–5 –1.7512 × 10–6 –2.1241 × 10–7 –2.8187 × 10–8
下载: 导出CSV
-
[1] Grevesse N, Sauval A J 1970 Astron. Astrophys. 9 232
[2] Douglas A E, Lutz B L 1970 Can. J. Phys. 48 247
Google Scholar
[3] Grevesse N, Sauval A J 1971a J. Quant. Spectrosc. Radiat. Transf. 11 65
Google Scholar
[4] Almeida A A, Sing P D 1978 Astrophys. Space Sci. 56 415
Google Scholar
[5] Gao W, Wang B B, Hu X J, Chai S, Han Y C, Greenwood J B 2017 Phys. Rev. A 96 013426
Google Scholar
[6] Wang B B, Han Y C, Gao W, Cong S L 2017 Phys. Chem. Chem. Phys. 19 22926
Google Scholar
[7] Moore P L, Browne J C, Matsen F A 1965 J. Chem. Phys. 43 903
Google Scholar
[8] Cosby P C, Helm H, Moseley J T 1980 Astrophys. J. 235 52
Google Scholar
[9] Barinovs G, Hemert M C V 2006 Astrophys. J. 636 923
Google Scholar
[10] Ram R S, Engleman R, Bernath P F 1998 J. Mol. Spectrosc. 190 341
Google Scholar
[11] Singh P D, Vanlandingham F G 1978 Astron. Astrophys. 66 87
Google Scholar
[12] Carlson T A, Copley J, Duric N, Elander N, Erman P, Larsson M, Lyyra M 1980 Astron. Astrophys. 83 238
[13] Hishikawa A, Karawajczyk A 1993 J. Mol. Spectrosc. 158 479
Google Scholar
[14] Davies P B, Martineau P M 1988 J. Chem. Phys. 88 485
Google Scholar
[15] Mosnier J P, Kennedy E T, Kampen P V, Cubaynes D, Guilbaud S, Sisourat N, Puglisi A, Carniato S, Bizau J M 2016 Phys. Rev. A 93 061401
Google Scholar
[16] Hirst D M 1986 Chem. Phys. Lett. 128 504
Google Scholar
[17] Langhoff S R, Davidson E R 1974 Int. J. Quantum Che. 8 61
Google Scholar
[18] Matos J M O, Kello V, Roos B O, Sadlej A J 1988 J. Chem. Phys. 89 423
Google Scholar
[19] Sannigrahi A B, Buenker R J, Hirsch G, Gu J P 1995 Chem. Phys. Lett. 237 204
Google Scholar
[20] Werner H J, Knowles P J, Lindh R, Manby F R, Schutz M, et al. Molpro, A Package of ab initio Programs (Version 2015.1) http://www.molpro.net [2021-03-08]
[21] Zhang Y G, Dou G, Cui J, Yu Y 2018 J. Mol. Struct. 1165 318
Google Scholar
[22] Neese F 2011 Wiley Interdisci. Rev. Comput. Mol. Sci. 2 73
[23] Biglari Z, Shayesteh A, Ershadifar S 2018 J. Quant. Spectrosc. Radiat. Transf. 221 80
Google Scholar
[24] Werner H J, Knowles P J, Lindh R, Manby F R, Schutz M, et al. Molpro, A Package of ab initio Programs (Version 2012.1) http://www.molpro.net [2021-03-08]
[25] Aguado A, Paniagua M 1992 J. Chem. Phys. 96 1265
Google Scholar
[26] Aguado A, Tablero C, Paniagua M 1998 Comput. Phys. Commun. 108 259
Google Scholar
[27] Varandas A J C 2007 J. Chem. Phys. 126 244105
Google Scholar
[28] Varandas A J C 2000 J. Chem. Phys. 113 8880
Google Scholar
[29] Jansen H B, Ross P 1969 Chem. Phys. Lett. 3 140
Google Scholar
[30] Liu B, McLean A D 1973 J. Chem. Phys. 59 4557
Google Scholar
[31] Karton A, Martin J M L 2006 Theor. Chem. ACC. 115 330
Google Scholar
[32] Yang C L, Huang Y J, Zhang X, Han K L 2003 J. Mol. Struc. Theochem. 625 289
Google Scholar
[33] Yang C L, Zhang X, Han K L 2004 J. Mol. Struc. Theochem. 676 209
Google Scholar
[34] Huber K P, Herzberg G 1979 Molecular Spectra and Molecular Structure (Vol. IV) (New York: Springer) p600
[35] Roy R J L 2017 J. Quant. Spectrosc. Radiat. Transf. 186 167
Google Scholar

 下载:
下载:








计量
- 文章访问数: 7130
- PDF下载量: 104
- 被引次数: 0
