










-
黑磷烯(black phosphorene, BP)因其“褶皱”的晶格结构而具有较高的比表面积, 在气体吸附及气体传感器方面应用具有很大的优势. 掺杂及缺陷对其传感性有较大的影响. 本文以基于密度泛函理论的第一性原理方法为基础探究了本征、Al掺杂、含P原子空缺以及P空位与Al掺杂共存的黑磷烯体系吸附甲醛前后的传感行为. 通过建立含缺陷和掺杂吸附体系的结构模型, 计算得出了吸附能、能带结构及电荷转移等电子结构参数. 结果表明, 本征BP烯以及含P原子空缺的BP烯体系对甲醛分子吸附能力较弱, P原子空缺对电导率以及电荷转移没有影响, 所以本征黑磷烯不适合用于传感器材料. Al掺杂和P空位与Al掺杂共存的BP烯体系, 吸附甲醛分子的能力明显比前两种情况增强, 电荷转移明显增加, 改变了载流子浓度, 提高了电导率. 此外, 在能带图中明显看到产生一个杂质能级, 有效带隙明显变窄, 表明Al掺杂提高了纯净和含P空位黑磷烯的传感性. 因此, Al掺杂和P空位与Al掺杂共存的BP烯体系预计可成为一种新的传感器材料.Black phosphorene (BP) has a high specific surface area due to its puckered honeycomb lattice structure, so it has great advantages in gas sensor applications. Doping and defects have a great effect on its sensitivity. Our aim is to obtain an insight into the sensing mechanism of black phosphorene towards CH2O, a hazardous organic compound. Based on the first-principles method of density functional theory (DFT), the sensing behaviors of the BP system, with intrinsic, Al doped, P vacancy-defected and P-vacancy and Al doping coexistent, before and after CH2O adsorption are studied. By establishing the structural models of four BP systems, the values of adsorption energy, energy band structure and charge transfer are calculated. Calculation results show that CH2O molecule prefers to be adsorbed perpendicular to the P vacancy-defected BP nanosheet with oxygen atom on the top site and close to the sheet. For the intrinsic, Al doped, P-vacancy and Al doping coexisting BP nanosheet, the CH2O molecule tilts towards the sheet surface. It is found that the CH2O adsorption on intrinsic BP nanosheet (adsorption energy is 0.179 eV) is very weak. In contrast, the adsorption of CH2O to the BP systems, with P vacancy-defected BP, Al doped, P-vacancy and Al doping coexistent, shows relatively high affinity (0.875, 0.542, 0.824 eV). Thus, Al doping, P vacancy or P-vacancy and Al-doping coexistence can substantially improve the adsorption ability of BP systems towards CH2O. In order to investigate the sensing mechanism of BP systems, the electronic properties such as the density of states, energy band and charge transfer are calculated. The change of energy gap of intrinsic BP nanosheet before and after CH2O adsorption is 0.024 eV, and that for P vacancy-defected BP nanosheet is zero. In addition, P atom vacancy has no effect on charge transfer. These suggest that the conductivity of intrinsic BP or P vacancy-defected BP nanosheet has not obviously changed, thereby, they are not suitable for sensor materials. For the BP system with Al doping or the coexistence of P vacancy and Al doping, it is obviously seen that an impurity level is generated in the energy band diagram, the effective band gap is significantly narrowed, indicating that the Al doping improves the sensitivity of BP. In addition, the charge transfer is significantly increased, which changes the carrier concentration and improves the electrical conductivity. Therefore, the BP system with Al doping or the coexistence of P vacancy and Al doping is expected to become a kind of new sensor material.
-
Keywords:
- black phosphorene /
- the first-principles method /
- formaldehyde /
- sensing behaviors
[1] Su S, Wu W H, Gao J M, Lu J X, Fang C H 2012 J. Mater. Chem. 22 18101
 Google Scholar
Google Scholar
[2] Schwierz F, Pezoldt J, Granzner R 2015 Nanoscale 7 8261
Google Scholar
[3] Perkins F K, Friedman A L, Cobas E, Campbell P M, Jernigan G G, Jonker B T 2013 Nano. Lett. 13 668
Google Scholar
[4] Schedin F, Geim A K, Morozov S V, Hill E W, Blake P, Katsnelson M I, Novoselov K S 2007 Nat. Mater. 6 652
Google Scholar
[5] 陈浩, 彭同江, 刘波, 孙红娟, 雷德会 2017 物理学报 66 080701
Google Scholar
Chen H, Peng T J, Liu B, Sun H J, Lei D H 2017 Acta Phys. Sin. 66 080701
Google Scholar
[6] 孙建平, 缪应蒙, 曹相春 2013 物理学报 62 036301
Google Scholar
Sun J P, Liao Y M, Cao X C 2013 Acta Phys. Sin. 62 036301
Google Scholar
[7] Samadizadeh M, Peyghan A A, Rastegar S F 2015 Chin. Chem. Lett. 26 1042
Google Scholar
[8] Liu S Y, Jiao X Q, Zhang G Y 2019 Chem. Phys. Lett. 726 77
Google Scholar
[9] Li L C, Yu Y J, Ye G J, Ge Q Q, Ou X D, Wu H, Fang D L, Chen X H, Zhang Y B 2014 Nat. Nanotechnol. 9 372
Google Scholar
[10] Yang A J, Wang D W, Wang X H, Zhang D Z, Koratkar N, Rong M Z 2018 Nano Today 20 13
Google Scholar
[11] Kou L Z, Frauenheim T, Chen C F 2014 Phys. Chem. Lett. 5 2675
Google Scholar
[12] Sun X L, Luan S, Shen H Y, Lei S Y 2018 Superlattices Microstruct. 124 168
Google Scholar
[13] 王晶儒, 岑超, 蔡绍洪 2016 贵州师范学院学报 32 17
Google Scholar
Wang J R, Cen C, Cai S H 2016 J. Guizhou Norm. Coll. 32 17
Google Scholar
[14] Xu Y, Gao W L 2018 J. Alloys and Compd. 737 365
Google Scholar
[15] 张国英, 焦兴强, 刘贵立 2020 沈阳师范大学学报 38 23
Google Scholar
Zhang G Y, Jiao X Q, Liu G L 2020 J. Shenyang Norm. Univ. (Nat. Sci. Ed.) 38 23
Google Scholar
[16] 原卫华, 毕世华, 曹茂盛 2015 材料导报 29 156
Google Scholar
Yuan W H, Bi S H, Cao M S 2015 Mate. Rep. 29 156
Google Scholar
[17] Segall M D, Lndan P L D, Probert M J, Pickard C J, Hasnip P J, Clark S J, Payne M C 2002 J. Phy. Condens. Matter 14 2717
Google Scholar
[18] Perdew J P, Chevary J A, Vosko S H, Jackson K A, Pederson M R, Singh D J, Fiolhais C 1992 Phys. Rev. B 46 6671
Google Scholar
[19] Qiao J S, Kong X H, Hu Z X, Yang F, Ji W 2014 Nat. Commun. 5 4475
Google Scholar
[20] Brown A, Rundqvist S 1965 Acta Cryst. 19 684
Google Scholar
[21] Fei R X, Yang L 2014 Nano Lett. 14 2884
Google Scholar
[22] Henkelmzn G, Arnaldsson A, Jonsson H 2006 Comput. Mater. Sci. 36 354
Google Scholar
[23] Peyghan A A, Hadipour N L, Bagheri Z 2013 J. Phys. Chem. C 117 2427
[24] Zhang Y H, Han L F, Xiao Y H, Jia D Z 2013 Comput. Mater. Sci. 69 222
Google Scholar
-
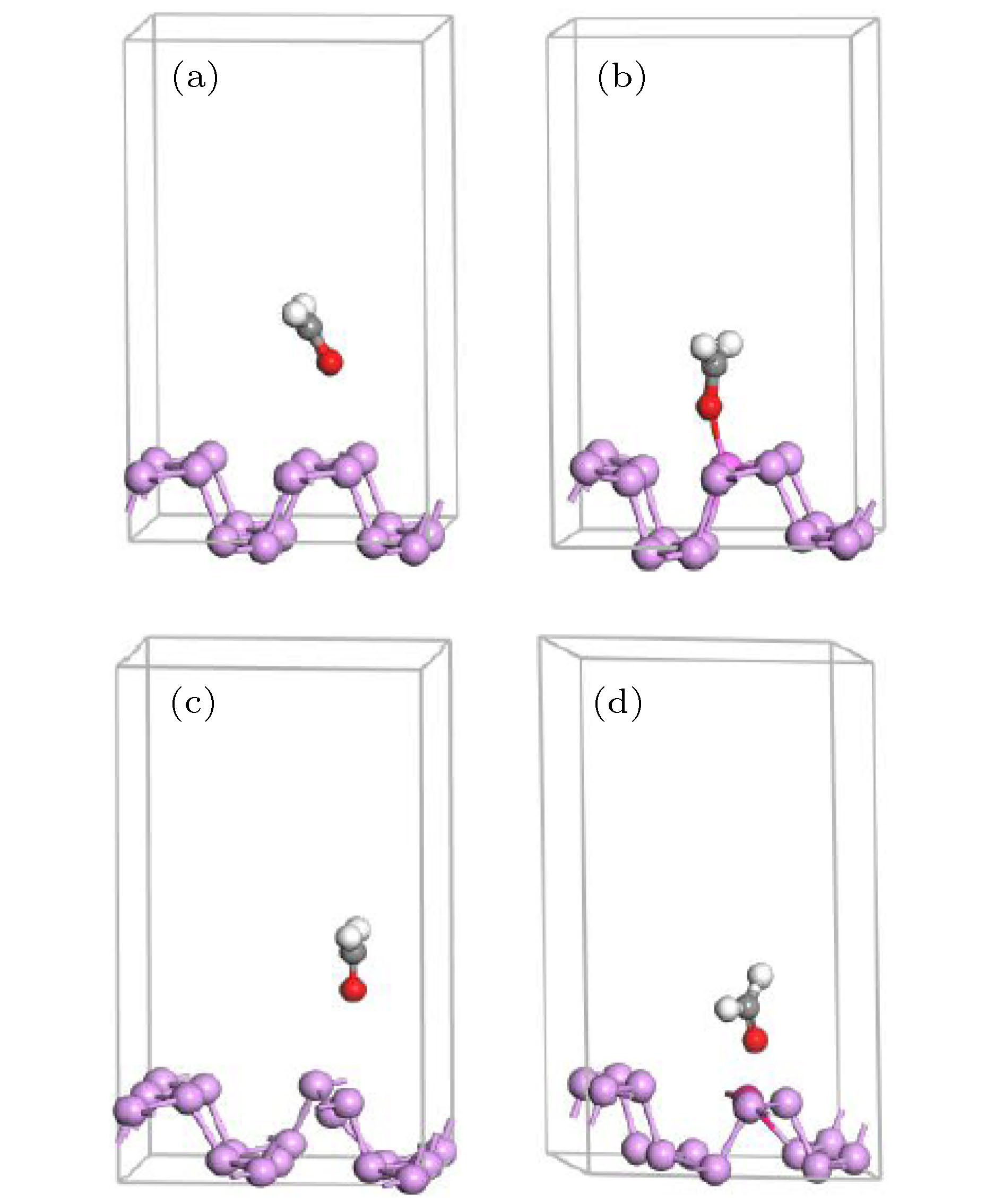
图 1 (a) 本征、(b) 掺杂Al、(c) 含P空位及(d) P空位与Al掺杂共存时BP烯甲醛吸附系统超原胞
Fig. 1. The supercells of CH2O/BP adsorption systems: (a) Intrinsic BP; (b) Al doped BP; (c) BP containing P vacancies; (d) BP with the coexistence of P-vacancies and Al doping.
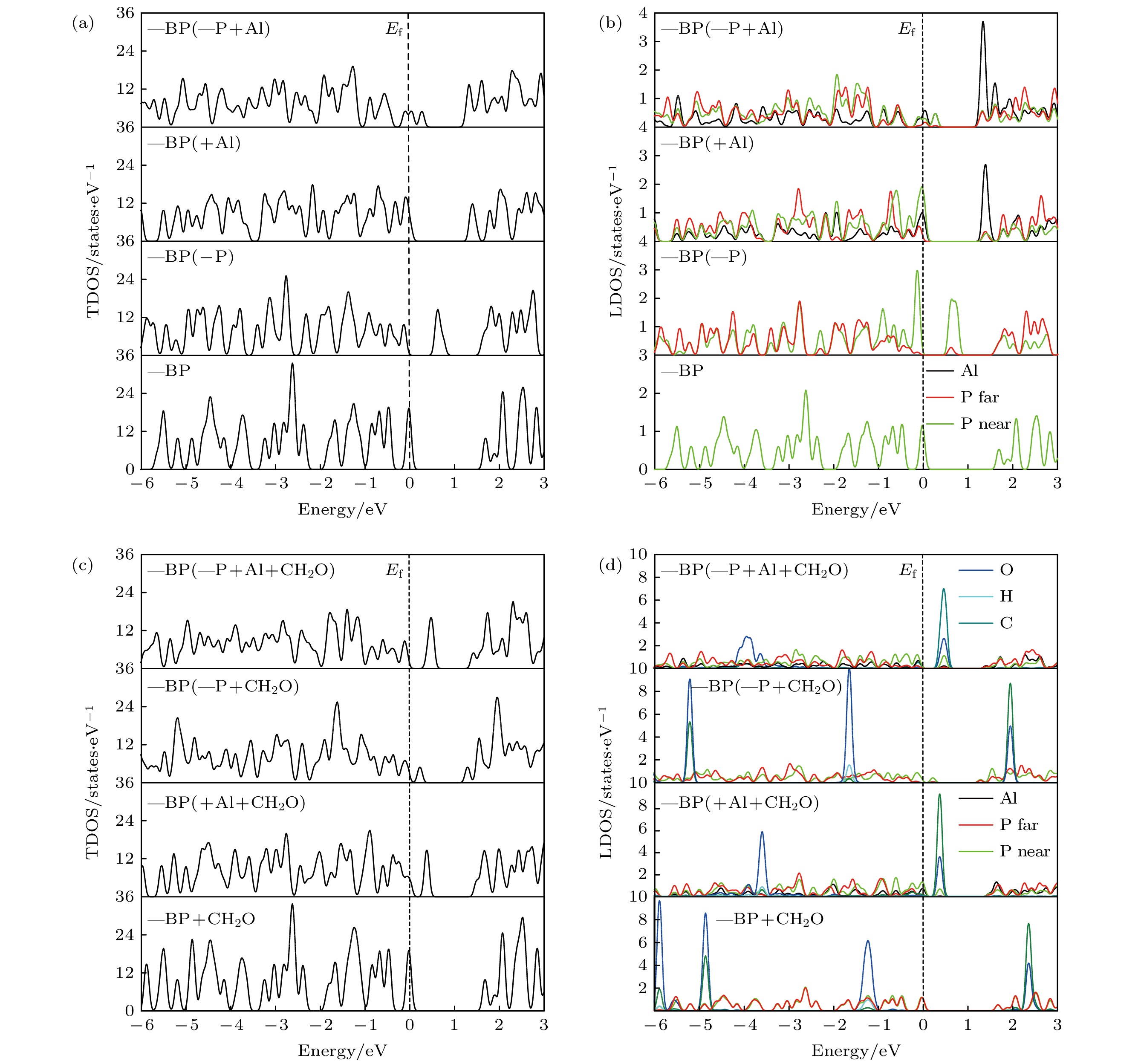
图 2 本征、Al掺杂、含P空位、P空位和Al掺杂共存时吸附或未吸附CH2O的BP态密度图 (a), (b) 各BP烯体系的总态密度和不同原子的局域态密度图; (c), (d) 各吸附CH2O的BP烯体系的总态密度和不同原子的局域态密度图
Fig. 2. The density of state of intrinsic, Al doped, P vacancy contained, P vacancy and Al doping coexisted BP with or without CH2O adsorption: (a), (b) The total density of state and the local density of state of different atoms of each BP system; (c), (d) the total density of state and the local density of state of different atoms of each BP system with CH2O adsorption.
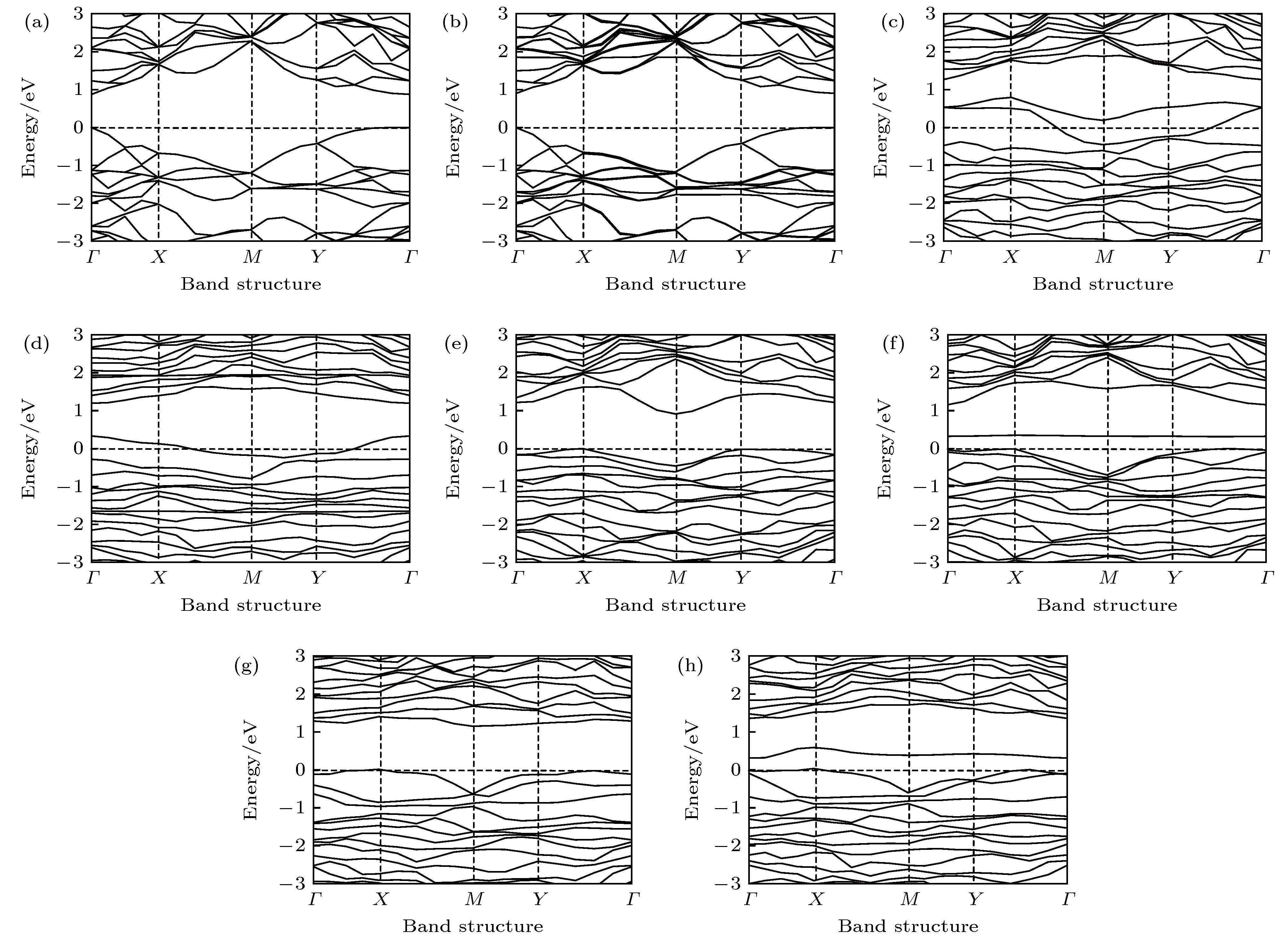
图 3 本征、Al掺杂、P空位、P空位和Al掺杂共存时吸附或未吸附CH2O的BP的能带图 (a) 本征BP烯; (b) 本征BP烯吸附CH2O; (c) 含P原子空位的BP烯; (d) 含P原子空位的BP烯吸附CH2O; (e) Al掺杂的BP烯; (f) Al掺杂BP烯吸附CH2O; (g) P空位和Al掺杂共存的BP烯; (h) P空位和Al掺杂共存BP烯吸附CH2O
Fig. 3. The energy band of intrinsic, Al doped, P vacancy contained, P vacancy and Al doping coexisted BP with or without CH2O adsorption: (a) Intrinsic BP; (b) intrinsic BP after adsorption of CH2O; (c) BP with P atom vacancy; (d) P atom vacancy contained BP after adsorption of CH2O; (e) Al doped BP; (f) Al doped BP after adsorption of CH2O; (g) P vacancy and Al doped coexisted BP; (h) P vacancy and Al doped coexisted BP after adsorption of CH2O.
表 1 考虑和不考虑自旋极化、取不同截断能和不同k点时计算的黑磷体材料的能隙和晶格参数
Table 1. Calculated energy gap and lattice parameters of bulk black phosphor using different cut-off energies and different k points considering or disregarding spin polarization.
截断能与k点/不加自旋 参数 Eg/ eV a/Å b/Å c/Å 300 eV/2 × 2 × 2 0.603 3.162 12.399 4.809 300 eV/3 × 3 × 3 0.358 3.348 11.195 4.549 320 eV/3 × 3 × 3 0.455 3.342 11.418 4.588 400 eV/3 × 3 × 3 0.490 3.340 11.466 4.598 300 eV/4 × 4 × 4 0.113 3.239 11.324 4.597 截断能与k点/加自旋 参数 Eg/eV a/Å b/Å c/Å 280 eV/3 × 3 × 3 0.178 3.350 11.137 4.485 300 eV 3 × 3 × 3 0.343 3.370 11.103 4.515 320 eV/3 × 3 × 3 0.239 3.346 11.277 4.501  下载: 导出CSV
下载: 导出CSV
表 2 BP或CH2O/BP中存在P空位、Al掺杂或P空位-Al掺杂对时的缺陷形成能
Table 2. Defect formation energies of P vacancy, Al impurity, or P-vacancy and Al impurity pair in BP or CH2O/BP.
Ef/eV AlP PV AlP + PV BP 0.256 2.325 0.878 CH2O/BP –0.107 1.631 0.234
下载: 导出CSV
表 3 本征、P空位、Al掺杂、P空位与Al掺杂共存时BP烯吸附甲醛的优化结构参数
Table 3. The optimal structure parameters of intrinsic, P-vacancy contained, Al doping, P-vacancy and Al doping coexisted BP systems adsorbed formaldehyde molecule.
基底 Ead/eV D(CH2O—BP) /Å L(C—O)/Å 本征 0.179 3.180 1.221 P空缺 0.875 4.007 1.223 Al掺杂 0.542 1.840 1.247 Al掺杂P空缺共存 0.824 1.886 1.265
下载: 导出CSV
表 4 甲醛分子及各原子的电荷得失
Table 4. The charge gain or loss of formaldehyde molecule and its atoms.
基底 电 荷/e H H C O ΔQ 本征 0.28 0.28 –0.06 –0.50 0 P空缺 0.27 0.27 –0.06 –0.48 0 Al掺杂 0.34 0.33 –0.13 –0.58 –0.04 Al掺杂与P空缺 0.31 0.34 –0.19 –0.61 –0.15
下载: 导出CSV
-
[1] Su S, Wu W H, Gao J M, Lu J X, Fang C H 2012 J. Mater. Chem. 22 18101
Google Scholar
[2] Schwierz F, Pezoldt J, Granzner R 2015 Nanoscale 7 8261
Google Scholar
[3] Perkins F K, Friedman A L, Cobas E, Campbell P M, Jernigan G G, Jonker B T 2013 Nano. Lett. 13 668
Google Scholar
[4] Schedin F, Geim A K, Morozov S V, Hill E W, Blake P, Katsnelson M I, Novoselov K S 2007 Nat. Mater. 6 652
Google Scholar
[5] 陈浩, 彭同江, 刘波, 孙红娟, 雷德会 2017 物理学报 66 080701
Google Scholar
Chen H, Peng T J, Liu B, Sun H J, Lei D H 2017 Acta Phys. Sin. 66 080701
Google Scholar
[6] 孙建平, 缪应蒙, 曹相春 2013 物理学报 62 036301
Google Scholar
Sun J P, Liao Y M, Cao X C 2013 Acta Phys. Sin. 62 036301
Google Scholar
[7] Samadizadeh M, Peyghan A A, Rastegar S F 2015 Chin. Chem. Lett. 26 1042
Google Scholar
[8] Liu S Y, Jiao X Q, Zhang G Y 2019 Chem. Phys. Lett. 726 77
Google Scholar
[9] Li L C, Yu Y J, Ye G J, Ge Q Q, Ou X D, Wu H, Fang D L, Chen X H, Zhang Y B 2014 Nat. Nanotechnol. 9 372
Google Scholar
[10] Yang A J, Wang D W, Wang X H, Zhang D Z, Koratkar N, Rong M Z 2018 Nano Today 20 13
Google Scholar
[11] Kou L Z, Frauenheim T, Chen C F 2014 Phys. Chem. Lett. 5 2675
Google Scholar
[12] Sun X L, Luan S, Shen H Y, Lei S Y 2018 Superlattices Microstruct. 124 168
Google Scholar
[13] 王晶儒, 岑超, 蔡绍洪 2016 贵州师范学院学报 32 17
Google Scholar
Wang J R, Cen C, Cai S H 2016 J. Guizhou Norm. Coll. 32 17
Google Scholar
[14] Xu Y, Gao W L 2018 J. Alloys and Compd. 737 365
Google Scholar
[15] 张国英, 焦兴强, 刘贵立 2020 沈阳师范大学学报 38 23
Google Scholar
Zhang G Y, Jiao X Q, Liu G L 2020 J. Shenyang Norm. Univ. (Nat. Sci. Ed.) 38 23
Google Scholar
[16] 原卫华, 毕世华, 曹茂盛 2015 材料导报 29 156
Google Scholar
Yuan W H, Bi S H, Cao M S 2015 Mate. Rep. 29 156
Google Scholar
[17] Segall M D, Lndan P L D, Probert M J, Pickard C J, Hasnip P J, Clark S J, Payne M C 2002 J. Phy. Condens. Matter 14 2717
Google Scholar
[18] Perdew J P, Chevary J A, Vosko S H, Jackson K A, Pederson M R, Singh D J, Fiolhais C 1992 Phys. Rev. B 46 6671
Google Scholar
[19] Qiao J S, Kong X H, Hu Z X, Yang F, Ji W 2014 Nat. Commun. 5 4475
Google Scholar
[20] Brown A, Rundqvist S 1965 Acta Cryst. 19 684
Google Scholar
[21] Fei R X, Yang L 2014 Nano Lett. 14 2884
Google Scholar
[22] Henkelmzn G, Arnaldsson A, Jonsson H 2006 Comput. Mater. Sci. 36 354
Google Scholar
[23] Peyghan A A, Hadipour N L, Bagheri Z 2013 J. Phys. Chem. C 117 2427
[24] Zhang Y H, Han L F, Xiao Y H, Jia D Z 2013 Comput. Mater. Sci. 69 222
Google Scholar

 下载:
下载:
计量
- 文章访问数: 9079
- PDF下载量: 219
- 被引次数: 0
