










-
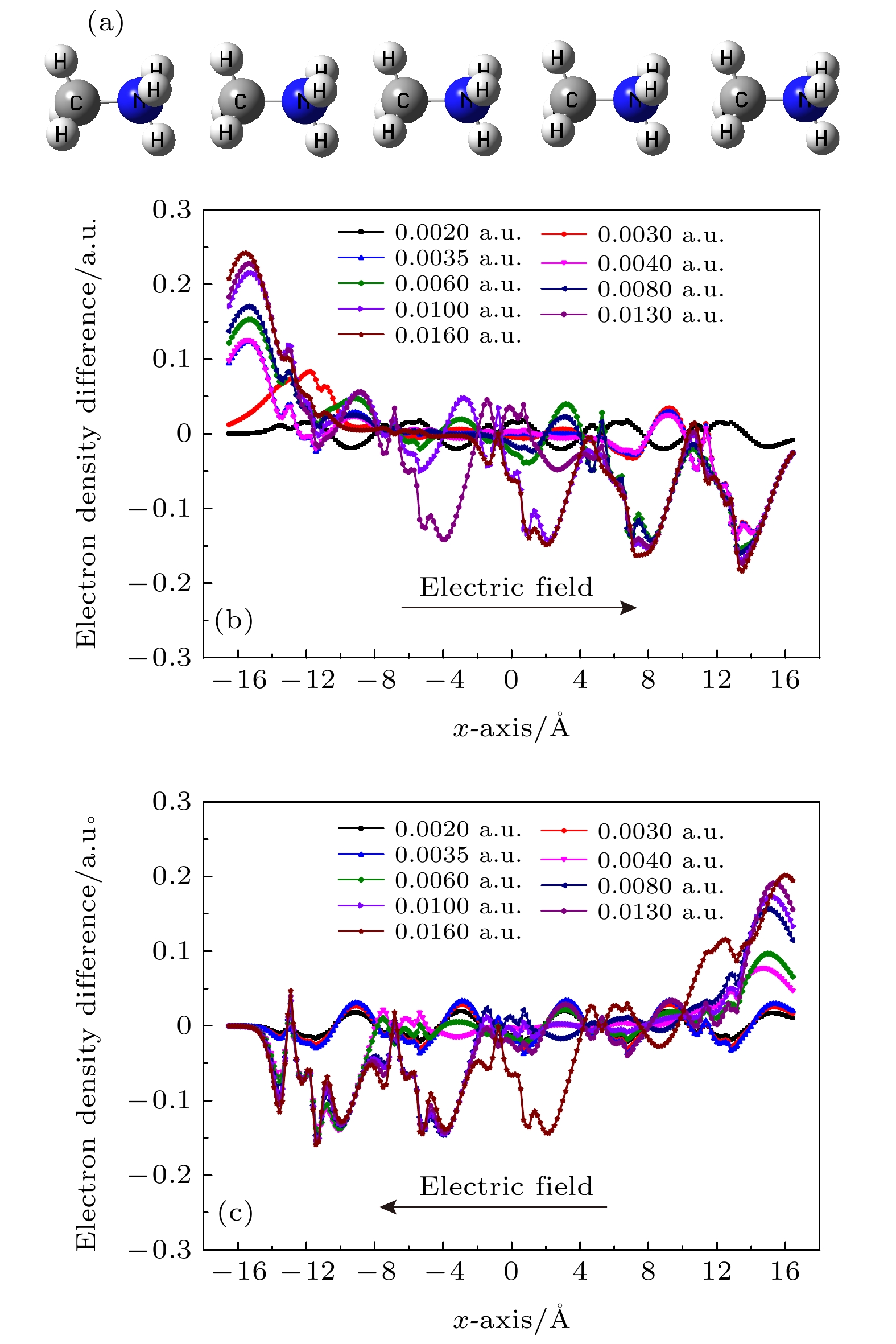
采用第一性原理在MP2/aug-cc-PVTZ 水平下优化得到CH3NH3多聚体的几何构型, 发现多聚体中CH3NH3沿C-N轴取向, 多聚体随着CH3NH3数量增加有收缩趋势, 这有利于无机框架的结构稳定, 多聚体的总偶极矩随着CH3NH3数量线性增加, 这导致了CH3NH3PbI3异质结的强极化. CH3NH3多聚体中未配对电子分布在每个CH3NH3的NH3-端, 轨道能量在–4.4—–3.2 eV之间. 计算静电势矢量场发现CH3NH
${}_3^+ $ 具有强亲电性, NH3-端比CH3-端有更强的亲电性, CH3NH3单体和CH3NH3多聚体具有弱亲电性和亲核性, CH3NH3多聚体的形成有效地减少CH3NH3与无机[PbI3]–框架之间的非谐振声子振动模式, 这有利于提高CH3NH3PbI3异质结中载流子传输. 电场作用下CH3NH3五聚体中未配对电子通过量子跃迁机制沿着C-N轴发生转移, 施加不同方向电场电子的转移效率是不一样的, 转移电子数量随着电场强度增加而增加, 通过这样的跃迁机制在外电场作用下电子很容易注入CH3NH3PbI3形成CH3NH3多聚体. 这些计算结果将有助于更深刻地理解有机-无机杂化钙钛矿太阳能电池高光电转换效率的根源.-
关键词:
- 有机-无机杂化钙钛矿 /
- 太阳能电池 /
- 第一性原理计算
CH3NH3PbI3 is one of the most promising candidates for high-performance hybrid organic-inorganic perovskite solar cells. The CH3NH3PbI3 single crystal and polycrystalline thin film exhibit the unique features of long carrier lifetimes and diffusion lengths, however, their carrier mobilities are in fact rather modest in a range from 1 cm2·V–1·s–1 to 100 cm2·V–1·s–1. Experimentally, the temperature dependence of mobility is described as T–1.3 to T–1.6 due to the acoustic phonon scattering. To be sure, the rotating CH3NH${}_3^+ $ cations are disadvantageous to the carrier transport and performance for CH3NH3PbI3 solar cells. The effect of the rotating CH3NH${}_3^+ $ cations on high-performance CH3NH3PbI3 solar cells remains an open question. The Gaussian 09 software has been utilized to optimize the geometrical structures of CH3NH3 dimer, trimer, tetramer, and pentamer in isolated state at the MP2 level with using the cc-PVTZ basis set. For CH3NH3 polymer, the mean distance between two centroids of neighboring CH3NH3 decreasing with the number of CH3NH3 is slightly smaller than the lattice constant 6.28 Å of tetragonal CH3NH3PbI3, which is advantageous to structural stability and higher structural order of inorganic [PbI3]– framework. It signifies that the long range order of electrically neutral CH3NH3 is easily formed for room-temperature CH3NH3PbI3. The total dipole moment linearly increases with the number of CH3NH3 for CH3NH3 polymer, and attains a large value 19.7 Debye for CH3NH3 pentamer, which may be the origin of strong polarization in CH3NH3PbI3 heterojunction. The molecular orbitals of five unpaired electrons for CH3NH3 pentamer are distributed around NH3-sides of five different CH3NH3 pentamers respectively, and these orbital energies are in a range from –4.4 eV to –3.2 eV. The unpaired electrons in CH3NH3 polymer have an electrostatic attraction on the CH3-side of neighboring CH3NH3, which is the key cause of forming the ordered CH3NH3 polymer. Hence it can be inferred that the orbital energies of unpaired electrons are getting closer when the longer range order of CH3NH3 are formed in room-temperature CH3NH3PbI3 through the interfacial electron injection. The vector field map of electrostatic potential (ESP) shows that CH3NH${}_3^+ $ has strong electrophilic character, and the NH3-side has a stronger electrophilic character than CH3-side, however, CH3NH3 monomer and polymer have weak electrophilic and nucleophilic character. Thus, the forming of CH3NH3 polymer at the CH3NH3PbI3 heterojunction leads the organic and inorganic portions to be decoupled, which can effectively reduce the anharmonic phonon modes. Under an applied electric field, the unpaired electrons in CH3NH3 pentamer can transfer along the C-N axis through the hopping mechanism. According to these results, we can draw three useful conclusions below. i) The electrons under an applied electric field are easily injected into the CH3NH3PbI3 material through the heterojunction, the CH3NH3 polymer is easily formed, and the unpaired electrons in polymer are transferred between two neighboring CH3NH3 through hopping mechanism. ii) The decoupling between organic CH3NH3 and inorganic [PbI3]– framework can effectively reduce the anharmonic phonon modes, which can lead the carrier scattering decrease and the efficiency of carrier separation and transport to improve; iii) The ordered CH3NH3 polymer at the CH3NH3PbI3 heterojunction can enhance the order of inorganic [PbI3]– framework. Our researches may help to further understand the origin of high power conversion efficiency (PCE) for hybrid organic-inorganic perovskite solar cells.[1] Zhang W, Eperon G E, Snaith H J 2016 Nat. Energy 1 16048
 Google Scholar
Google Scholar
[2] Kojima A, Teshima K, Shirai Y, Miyasaka T 2009 J. Am. Chem. Soc. 131 6050
Google Scholar
[3] Ding H, Li B, Zareen S, Li G, Tu Y, Zhang D, Cao X, Xu Q, Yang S, Tait S L, Zhu J 2020 ACS Appl. Mater. Interfaces 12 28861
Google Scholar
[4] Breternitz J, Lehmann F, Barnett S A, Nowell H, Schorr S 2020 Angew. Chem. Int. Ed. 59 424
Google Scholar
[5] Herz L M 2016 Annu. Rev. Phys. Chem. 67 65
Google Scholar
[6] Bi Y, Hutter E M, Fang Y, Dong Q, Huang J, Savenije T J 2016 J. Phys. Chem. Lett. 7 923
Google Scholar
[7] Ponseca C S, Savenije T J, Abdellah M, Zheng K, Yartsev A, Pascher T, Harlang T, Chabera P, Pullerits T, Stepanov A, Wolf J P, Sundströ V 2014 J. Am. Chem. Soc. 136 5189
Google Scholar
[8] Chen Y, Yi H T, Wu X, Haroldson R, Gartstein Y N, Rodionov Y I, Tikhonov K S, Zakhidov A, Zhu Z Y, Podzorov V 2016 Nat. Commun. 7 12253
Google Scholar
[9] He J L, Fang W H, Long R, Prezhdo O V 2020 J. Am. Chem. Soc. 142 14664
Google Scholar
[10] Li W W, Man Z Y, Zeng J T, Zheng L Y, Li G R, Kassiba A 2020 J. Phys. Chem. C 124 13348
Google Scholar
[11] Brenner T M, Egger D A, Rappe A M, Kronik L, Hodes G, Cahen D 2015 J. Phys. Chem. Lett. 6 4754
Google Scholar
[12] Dong Q, Fang Y, Shao Y, Mulligan P, Qiu J, Cao L, Huang J 2015 Science 347 967
Google Scholar
[13] Shi D, Adinolfi V, Comin R, Yuan M, Alarousu E, Buin A, Chen Y, Hoogland S, Rothenberger A, Katsiev K, Losovyj Y, Zhang X, Dowben P A, Mohammed O F, Sargent E H, Bakr O M 2015 Science 347 519
Google Scholar
[14] Mei Y, Zhang C, Vardeny Z V, Jurchescu O D 2015 MRS Commun. 5 297
Google Scholar
[15] Savenije T J, Ponseca C S, Kunneman L, Abdellah M, Zheng K, Tian Y, Zhu Q, Canton S E, Scheblykin I G, Pullerits T, Yartsev A, Sundstrom V 2014 J. Phys. Chem. Lett. 5 2189
Google Scholar
[16] Karakus M, Jensen S A, D’Angelo F, Turchinovich D, Bonn M, Canovas E 2015 J. Phys. Chem. Lett. 6 4991
Google Scholar
[17] Oga H, Saeki A, Ogomi Y, Hayase S, Seki S 2014 J. Am. Chem. Soc. 136 13818
Google Scholar
[18] He Y, Galli G 2014 Chem. Mater. 26 5394
Google Scholar
[19] Wang Y, Zhang Y, Zhang P, Zhang W 2015 Phys. Chem. Chem. Phys. 17 11516
Google Scholar
[20] Whalley L D, Skelton J M, Frost J M, Walsh A 2016 Phys. Rev. B 94 220301
Google Scholar
[21] Monahan D M, Guo L, Lin J, Dou L, Yang P, Fleming G R 2017 J. Phys. Chem. Lett. 8 83211
[22] Ma J, Wang L W 2017 Nano Lett. 17 3646
Google Scholar
[23] Frisch M J, Trucks G W, Schlegel H B, et al. 2009 Gaussian 09 (Revision C.01) (Wallingford: Gaussian, Inc.)
[24] Murray J S, Brinck T, Lane P, Paulsen K, Politzer P 1994 J. Mol. Struct. Theochem. 307 55
Google Scholar
[25] Lefebvre C, Rubez G, Khartabil H, Boisson J C, Contreras-García J, Hénon E 2017 Phys. Chem. Chem. Phys. 19 17928
Google Scholar
[26] Belpassi L, Infante I, Tarantelli F, Visscher L 2008 J. Am. Chem. Soc. 130 1048
Google Scholar
[27] Zhang A, Chen Y L, Zhang C X, Yan J 2019 Chin. Phys. Lett. 36 026701
Google Scholar
[28] 张翱, 陈云琳, 闫君, 张春秀 2018 物理学报 67 106701
Google Scholar
Zhang A, Chen Y L, Yan J, Zhang C X 2018 Acta Phys. Sin. 67 106701
Google Scholar
[29] 张翱, 张春秀, 陈云琳, 张春梅, 孟涛 2020 物理学报 69 118801
Google Scholar
Zhang A, Zhang C X, Chen Y L, Zhang C M, Meng T 2020 Acta Phys. Sin 69 118801
Google Scholar
-
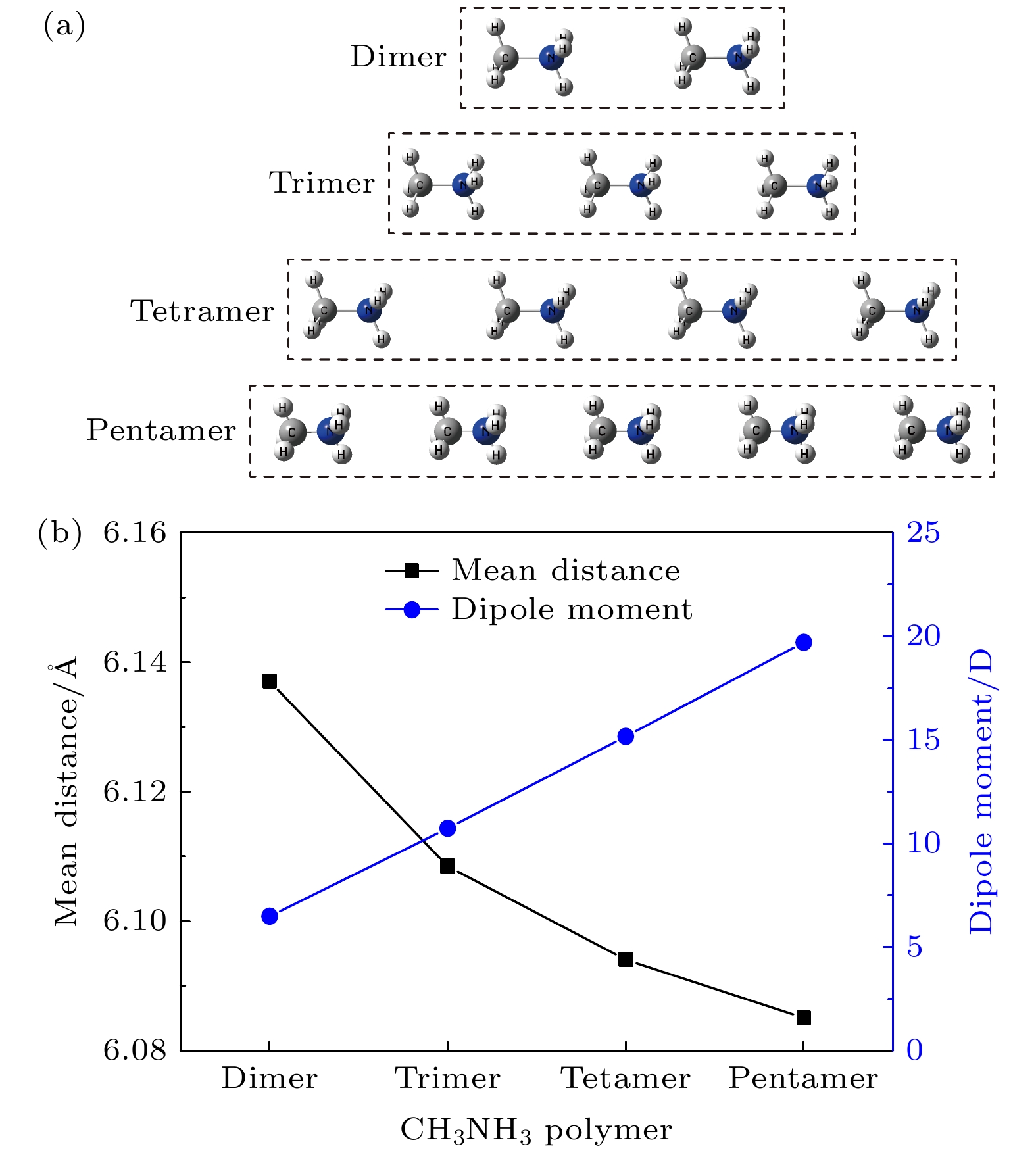
图 1 在MP2/Aug-cc-PVTZ水平下优化CH3NH3二聚体、三聚体、四聚体、五聚体的邻近两个分子质心之间的平均距离和电偶极矩
Fig. 1. Mean distance between two centroids of neighboring molecules and dipole moments for optimized CH3NH3 dimer, trimer, tetramer, and pentamer at MP2/Aug-cc-PVTZ level.
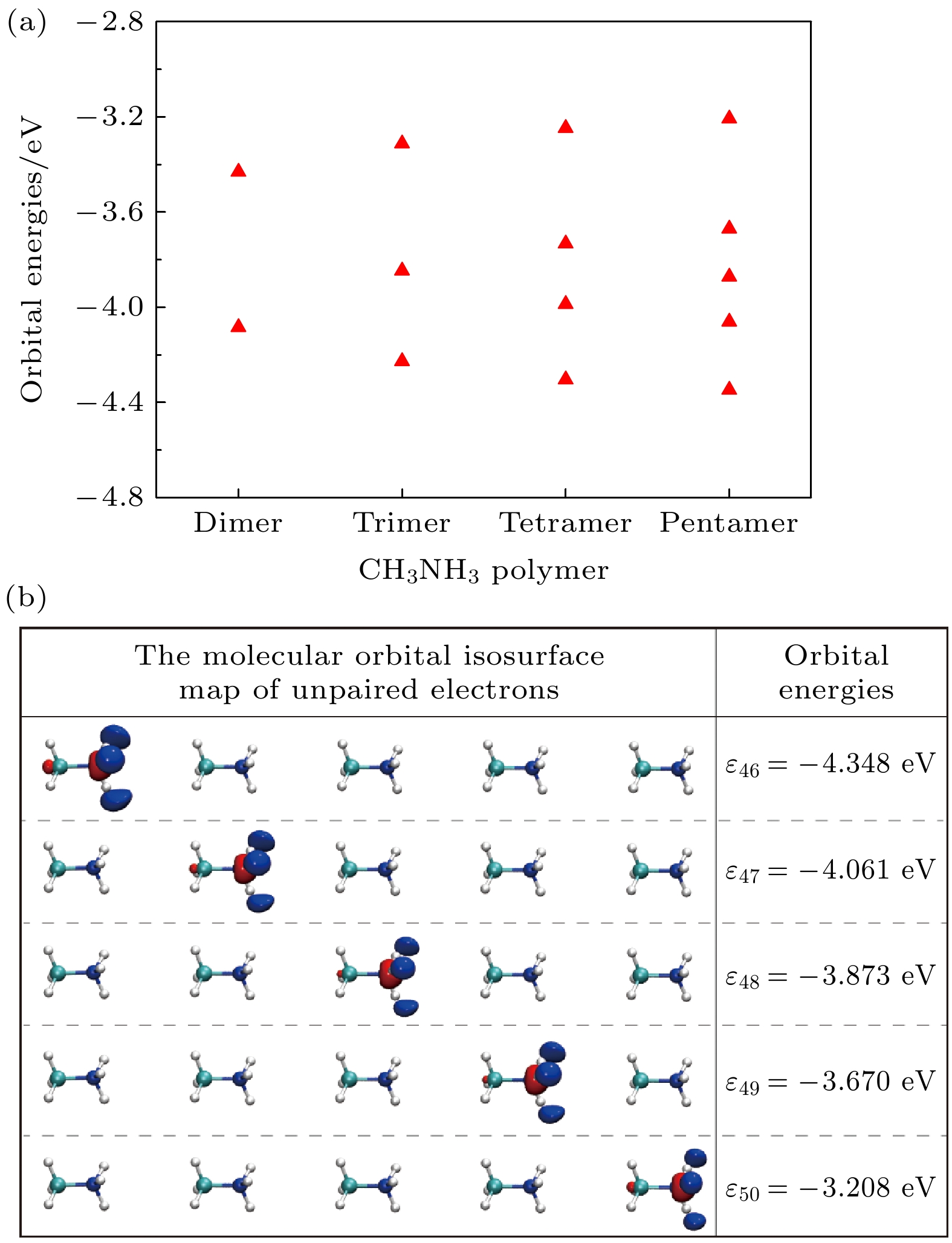
图 2 在MP2/Aug-cc-PVTZ水平下 (a)优化的CH3NH3多聚体未配对电子的分子轨道能, (b) 优化的CH3NH3五聚体的分子轨道等值面图和轨道能量, 红色和蓝色分别表示正相和负相
Fig. 2. (a) The molecular orbital energies of unpaired electrons for optimized CH3NH3 polymer, and (b) the molecular orbitals isosurface map and energies of unpaired electrons of optimized CH3NH3 pentamer at MP2/Aug-cc-PVTZ level. Red and blue colors correspond to positive and negative phases, respectively.
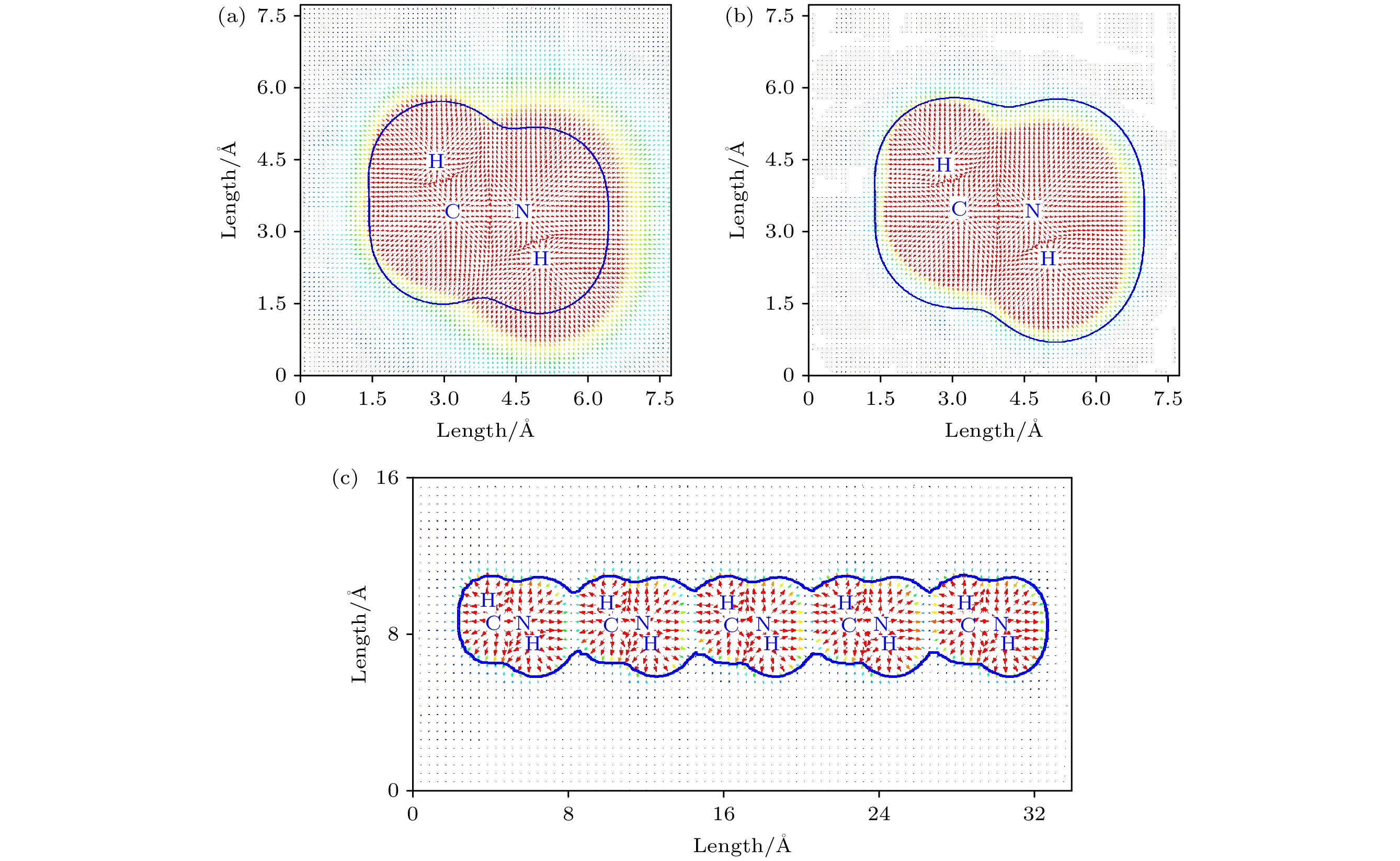
图 3 在MP2/Aug-cc-PVTZ水平下优化的 (a) CH3NH
${}_3^+ $ , (b) CH3NH3, (c) CH3NH3五聚体的静电势矢量场图, 蓝色的轮廓线表示范德瓦耳斯表面, 红色的箭头表示对应坐标处的电场Fig. 3. Vector field map of ESP for optimized (a) CH3NH
${}_3^+ $ , (b) CH3NH3, and (c) CH3NH3 pentamer at MP2/Aug-cc-PVTZ level. The blue contour line and red arrow represent van der Waals surface and electric field at corresponding position, respectively.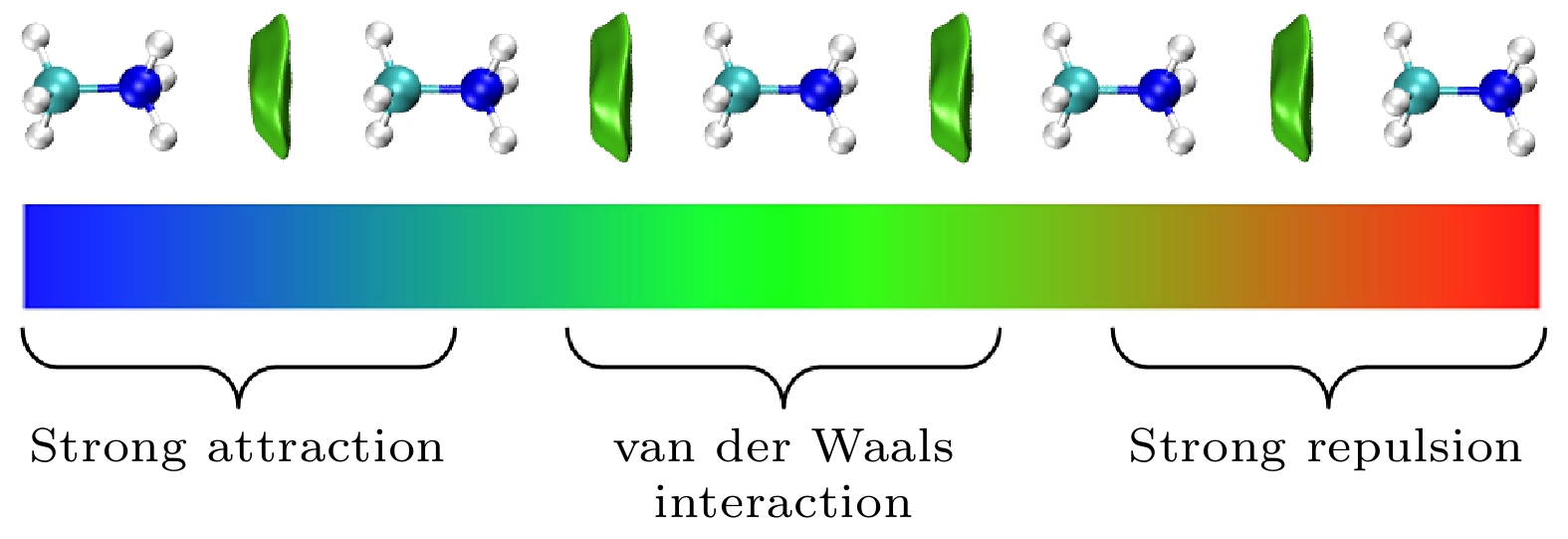
图 4 在MP2/Aug-cc-PVTZ水平下, 在优化的CH3NH3五聚体中通过RDG方法图形化CH3NH3之间的相互作用
Fig. 4. Weak interaction between two adjacent CH3NH3 in optimized CH3NH3 pentamer at MP2/Aug-cc-PVTZ level is visualized by RDG method.
-
[1] Zhang W, Eperon G E, Snaith H J 2016 Nat. Energy 1 16048
Google Scholar
[2] Kojima A, Teshima K, Shirai Y, Miyasaka T 2009 J. Am. Chem. Soc. 131 6050
Google Scholar
[3] Ding H, Li B, Zareen S, Li G, Tu Y, Zhang D, Cao X, Xu Q, Yang S, Tait S L, Zhu J 2020 ACS Appl. Mater. Interfaces 12 28861
Google Scholar
[4] Breternitz J, Lehmann F, Barnett S A, Nowell H, Schorr S 2020 Angew. Chem. Int. Ed. 59 424
Google Scholar
[5] Herz L M 2016 Annu. Rev. Phys. Chem. 67 65
Google Scholar
[6] Bi Y, Hutter E M, Fang Y, Dong Q, Huang J, Savenije T J 2016 J. Phys. Chem. Lett. 7 923
Google Scholar
[7] Ponseca C S, Savenije T J, Abdellah M, Zheng K, Yartsev A, Pascher T, Harlang T, Chabera P, Pullerits T, Stepanov A, Wolf J P, Sundströ V 2014 J. Am. Chem. Soc. 136 5189
Google Scholar
[8] Chen Y, Yi H T, Wu X, Haroldson R, Gartstein Y N, Rodionov Y I, Tikhonov K S, Zakhidov A, Zhu Z Y, Podzorov V 2016 Nat. Commun. 7 12253
Google Scholar
[9] He J L, Fang W H, Long R, Prezhdo O V 2020 J. Am. Chem. Soc. 142 14664
Google Scholar
[10] Li W W, Man Z Y, Zeng J T, Zheng L Y, Li G R, Kassiba A 2020 J. Phys. Chem. C 124 13348
Google Scholar
[11] Brenner T M, Egger D A, Rappe A M, Kronik L, Hodes G, Cahen D 2015 J. Phys. Chem. Lett. 6 4754
Google Scholar
[12] Dong Q, Fang Y, Shao Y, Mulligan P, Qiu J, Cao L, Huang J 2015 Science 347 967
Google Scholar
[13] Shi D, Adinolfi V, Comin R, Yuan M, Alarousu E, Buin A, Chen Y, Hoogland S, Rothenberger A, Katsiev K, Losovyj Y, Zhang X, Dowben P A, Mohammed O F, Sargent E H, Bakr O M 2015 Science 347 519
Google Scholar
[14] Mei Y, Zhang C, Vardeny Z V, Jurchescu O D 2015 MRS Commun. 5 297
Google Scholar
[15] Savenije T J, Ponseca C S, Kunneman L, Abdellah M, Zheng K, Tian Y, Zhu Q, Canton S E, Scheblykin I G, Pullerits T, Yartsev A, Sundstrom V 2014 J. Phys. Chem. Lett. 5 2189
Google Scholar
[16] Karakus M, Jensen S A, D’Angelo F, Turchinovich D, Bonn M, Canovas E 2015 J. Phys. Chem. Lett. 6 4991
Google Scholar
[17] Oga H, Saeki A, Ogomi Y, Hayase S, Seki S 2014 J. Am. Chem. Soc. 136 13818
Google Scholar
[18] He Y, Galli G 2014 Chem. Mater. 26 5394
Google Scholar
[19] Wang Y, Zhang Y, Zhang P, Zhang W 2015 Phys. Chem. Chem. Phys. 17 11516
Google Scholar
[20] Whalley L D, Skelton J M, Frost J M, Walsh A 2016 Phys. Rev. B 94 220301
Google Scholar
[21] Monahan D M, Guo L, Lin J, Dou L, Yang P, Fleming G R 2017 J. Phys. Chem. Lett. 8 83211
[22] Ma J, Wang L W 2017 Nano Lett. 17 3646
Google Scholar
[23] Frisch M J, Trucks G W, Schlegel H B, et al. 2009 Gaussian 09 (Revision C.01) (Wallingford: Gaussian, Inc.)
[24] Murray J S, Brinck T, Lane P, Paulsen K, Politzer P 1994 J. Mol. Struct. Theochem. 307 55
Google Scholar
[25] Lefebvre C, Rubez G, Khartabil H, Boisson J C, Contreras-García J, Hénon E 2017 Phys. Chem. Chem. Phys. 19 17928
Google Scholar
[26] Belpassi L, Infante I, Tarantelli F, Visscher L 2008 J. Am. Chem. Soc. 130 1048
Google Scholar
[27] Zhang A, Chen Y L, Zhang C X, Yan J 2019 Chin. Phys. Lett. 36 026701
Google Scholar
[28] 张翱, 陈云琳, 闫君, 张春秀 2018 物理学报 67 106701
Google Scholar
Zhang A, Chen Y L, Yan J, Zhang C X 2018 Acta Phys. Sin. 67 106701
Google Scholar
[29] 张翱, 张春秀, 陈云琳, 张春梅, 孟涛 2020 物理学报 69 118801
Google Scholar
Zhang A, Zhang C X, Chen Y L, Zhang C M, Meng T 2020 Acta Phys. Sin 69 118801
Google Scholar



 下载:
下载:


计量
- 文章访问数: 7115
- PDF下载量: 94
- 被引次数: 0
