










-
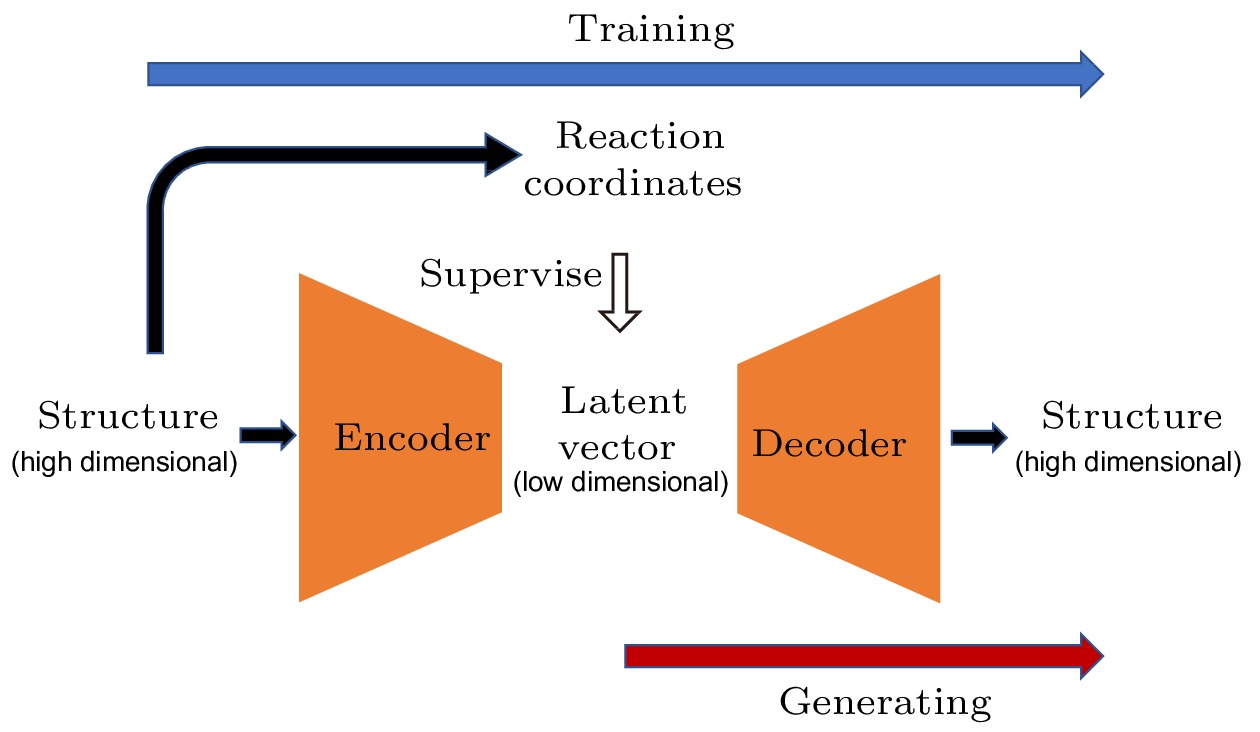
蛋白质的功能往往与其结构和动态变化密切相关. 分子动力学模拟是研究蛋白质结构变化的有效方法, 然而使用分子动力学模拟对蛋白质的构象空间进行采样需要花费很长的时间. 近年来的一些研究表明, 使用简单的机器学习模型——自编码器及其改进型, 可以在有限采样的情况下, 快速完成对蛋白质构象空间的探索. 该模型通过训练神经网络, 完成对隐变量的提取, 同时根据其产生构象, 但是由于提取出的隐变量没有直观的含义, 探索构象空间的方向会受到影响. 本工作通过引入反应坐标(如质心距离等), 建立了一个中间层受监督的自编码器模型, 以解决上述问题. 该模型应用于噬菌体T4溶菌酶和腺苷酸激酶两个蛋白质分子, 结果表明, 仅使用短时间分子动力学模拟作为训练数据, 就可以探索到这两种蛋白分子的多种典型构象. 有监督(合理的反应坐标或者实验数据等)的自编码器模型有望成为探索蛋白质构象空间的有效工具.Protein function is related to its structure and dynamic change. Molecular dynamics simulation is an important tool for studying protein dynamics by exploring its conformational space, however, conformational sampling is a nontrivial issue, because of the risk of missing key details during sampling. In recent years, deep learning methods, such as auto-encoder, can couple with MD to explore conformational space of protein. After being trained with the MD trajectories, auto-encoder can generate new conformations quickly by inputting random numbers in low dimension space. However, some problems still exist, such as requirements for the quality of the training set, the limitation of explorable area and the undefined sampling direction. In this work, we build a supervised auto-encoder, in which some reaction coordinates are used to guide conformational exploration along certain directions. We also try to expand the explorable area by training through the data generated by the model. Two multi-domain proteins, bacteriophage T4 lysozyme and adenylate kinase, are used to illustrate the method. In the case of the training set consisting of only under-sampled simulated trajectories, the supervised auto-encoder can still explore along the given reaction coordinates. The explored conformational space can cover all the experimental structures of the proteins and be extended to regions far from the training sets. Having been verified by molecular dynamics and secondary structure calculations, most of the conformations explored are found to be plausible. The supervised auto-encoder provides a way to efficiently expand the conformational space of a protein with limited computational resources, although some suitable reaction coordinates are required. By integrating appropriate reaction coordinates or experimental data, the supervised auto-encoder may serve as an efficient tool for exploring conformational space of proteins.
-
Keywords:
- protein conformational space /
- molecular dynamics simulation /
- machine learning /
- auto-encoder
[1] Chu X, Gan L, Wang E, Wang J 2013 Proc. Natl. Acad. Sci. U.S.A. 110 E2342
 Google Scholar
Google Scholar
[2] Smyth M S, Martin J H 2000 Mol. Pathol. 53 8
Google Scholar
[3] Danev R, Yanagisawa H, Kikkawa M 2019 Trends Biochem. Sci. 44 837
Google Scholar
[4] Vincenzi M, Mercurio F A, Leone M 2021 Curr. Med. Chem. 28 2729
Google Scholar
[5] Kachala M, Valentini E, Svergun D I 2015 Adv. Exp. Med. Biol. 870 261
Google Scholar
[6] Chu F, Thornton D T, Nguyen H T 2018 Methods 144 53
Google Scholar
[7] Bhaumik S R 2021 Emerg. Top Life Sci. 5 49
Google Scholar
[8] Jumper J, Evans R, Pritzel A, Green T, Figurnov M, Ronneberger O, Tunyasuvunakool K, Bates R, Žídek A, Potapenko A, Bridgland A, Meyer C, Kohl S A A, Ballard A J, Cowie A, Romera-Paredes B, Nikolov S, Jain R, Adler J, Back T, Petersen S, Reiman D, Clancy E, Zielinski M, Steinegger M, Pacholska M, Berghammer T, Bodenstein S, Silver D, Vinyals O, Senior A W, Kavukcuoglu K, Kohli P, Hassabis D 2021 Nature 596 583
Google Scholar
[9] Baek M, DiMaio F, Anishchenko I, Dauparas J, Ovchinnikov S, Lee G R, Wang J, Cong Q, Kinch L N, Schaeffer R D, Millán C, Park H, Adams C, Glassman C R, DeGiovanni A, Pereira J H, Rodrigues A V, van Dijk A A, Ebrecht A C, Opperman D J, Sagmeister T, Buhlheller C, Pavkov-Keller T, Rathinaswamy M K, Dalwadi U, Yip C K, Burke J E, Garcia K C, Grishin N V, Adams P D, Read R J, Baker D 2021 Science 373 871
Google Scholar
[10] Karplus M, Kuriyan J 2005 Proc. Natl. Acad. Sci. 102 6679
Google Scholar
[11] Bernardi R C, Melo M C R, Schulten K 2015 Biochim. Biophys. Acta 1850 872
Google Scholar
[12] Mu J, Liu H, Zhang J, Luo R, Chen H F 2021 J. Chem. Inf. Model. 61 1037
Google Scholar
[13] Lemke T, Peter C 2019 J. Chem. Theory Comput. 15 1209
Google Scholar
[14] Zhu J, Wang J, Han W, Xu D 2022 Nat. Commun. 13 1661
Google Scholar
[15] Hinton G E, Salakhutdinov R R 2006 Science 313 504
Google Scholar
[16] Degiacomi M T 2019 Structure 27 1034
Google Scholar
[17] Wen B, Peng J, Zuo X, Gong Q, Zhang Z 2014 Biophysical J. 107 956
Google Scholar
[18] Giri Rao V V H, Gosavi S 2014 PLOS Computational Biology 10 e1003938
Google Scholar
[19] Abraham M J, Murtola T, Schulz R, Páll S, Smith J C, Hess B, Lindahl E 2015 SoftwareX 1–2 19
Google Scholar
[20] Weaver L H, Matthews B W 1987 J. Mol. Biol. 193 189
Google Scholar
[21] Zhang X J, Wozniak J A, Matthews B W 1995 J. Mol. Biol. 250 527
Google Scholar
[22] Müller C W, Schulz G E 1992 J. Mol. Biol. 224 159
Google Scholar
[23] Müller C W, Schlauderer G J, Reinstein J, Schulz G E 1996 Structure 4 147
Google Scholar
[24] Hornak V, Abel R, Okur A, Strockbine B, Roitberg A, Simmerling C 2006 Proteins Struct. Funct. Bioinf. 65 712
Google Scholar
[25] Izadi S, Anandakrishnan R, Onufriev A V 2014 J. Phys. Chem. Lett. 5 3863
Google Scholar
[26] Huang J, Rauscher S, Nawrocki G, Ran T, Feig M, de Groot B L, Grubmüller H, MacKerell A D 2017 Nat. Methods 14 71
Google Scholar
[27] Bussi G, Donadio D, Parrinello M 2007 J. Chem. Phys. 126 014101
Google Scholar
[28] Essmann U, Perera L E, Berkowitz M L, Darden T A, Lee H C, Pedersen L G 1995 J. Chem. Phys. 103 8577
Google Scholar
[29] Kingma D P, Ba J 2014 arXiv:1412.6980 [cs.LG
[30] Lovell S C, Davis I W, Arendall III W B, de Bakker P I W, Word J M, Prisant M G, Richardson J S, Richardson D C 2003 Proteins Struct. Funct. Bioinf. 50 437
Google Scholar
[31] Eastman P, Swails J, Chodera J D, McGibbon R T, Zhao Y, Beauchamp K A, Wang L P, Simmonett A C, Harrigan M P, Stern C D, Wiewiora R P, Brooks B R, Pande V S 2017 PLoS Comput. Biol. 13 e1005659
Google Scholar
[32] Shirts M R, Klein C, Swails J M, Yin J, Gilson M K, Mobley D L, Case D A, Zhong E D 2017 J. Comput. -Aided Mol. Des. 31 147
Google Scholar
[33] Touw W G, Baakman C, Black J, te Beek T A, Krieger E, Joosten R P, Vriend G 2015 Nucleic Acids Res. 43 D364
Google Scholar
-
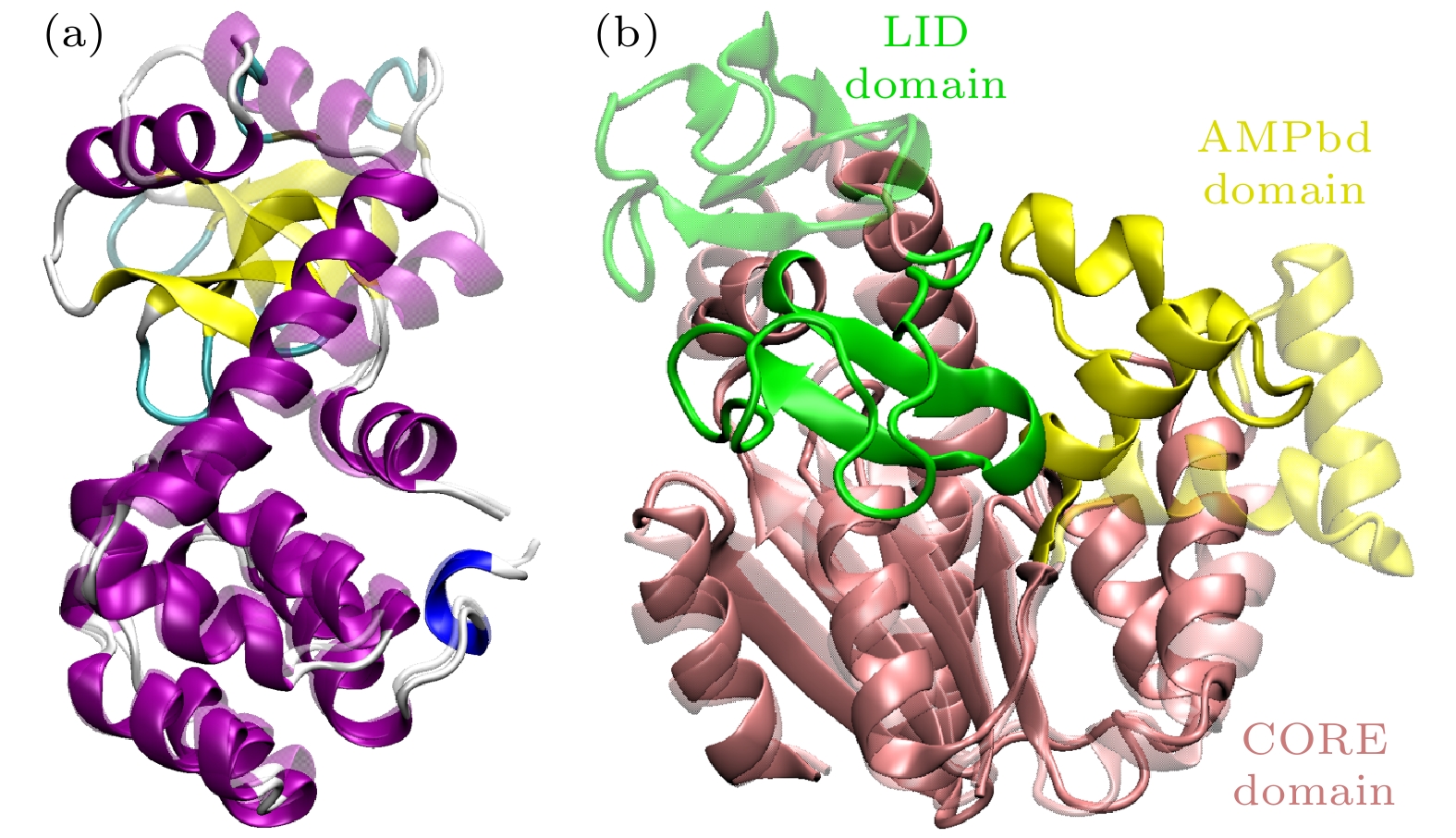
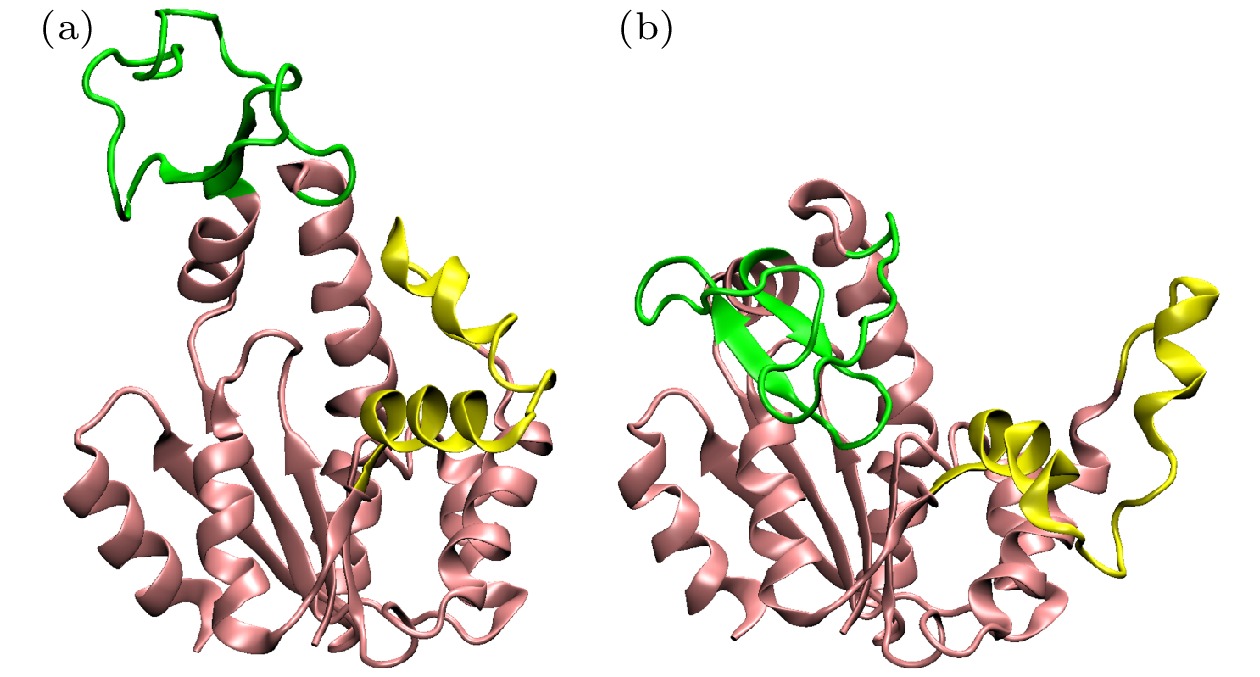
图 2 本研究中使用的两种蛋白质分子的不同结构 (a) T4L的闭合(不透明)和打开(透明)结构, 紫色为α螺旋, 黄色为β折叠; (b) AdK的闭合(不透明)和打开(透明)结构, 不同颜色表示不同的结构域
Fig. 2. Different structures of the two proteins in the work. (a) The close (opaque) and open (transparent) state of T4L. α-helix is colored in purple and β-sheet is colored in yellow. (b) The close (opaque) and open (transparent) state of AdK. Different domains are colored in different colors.

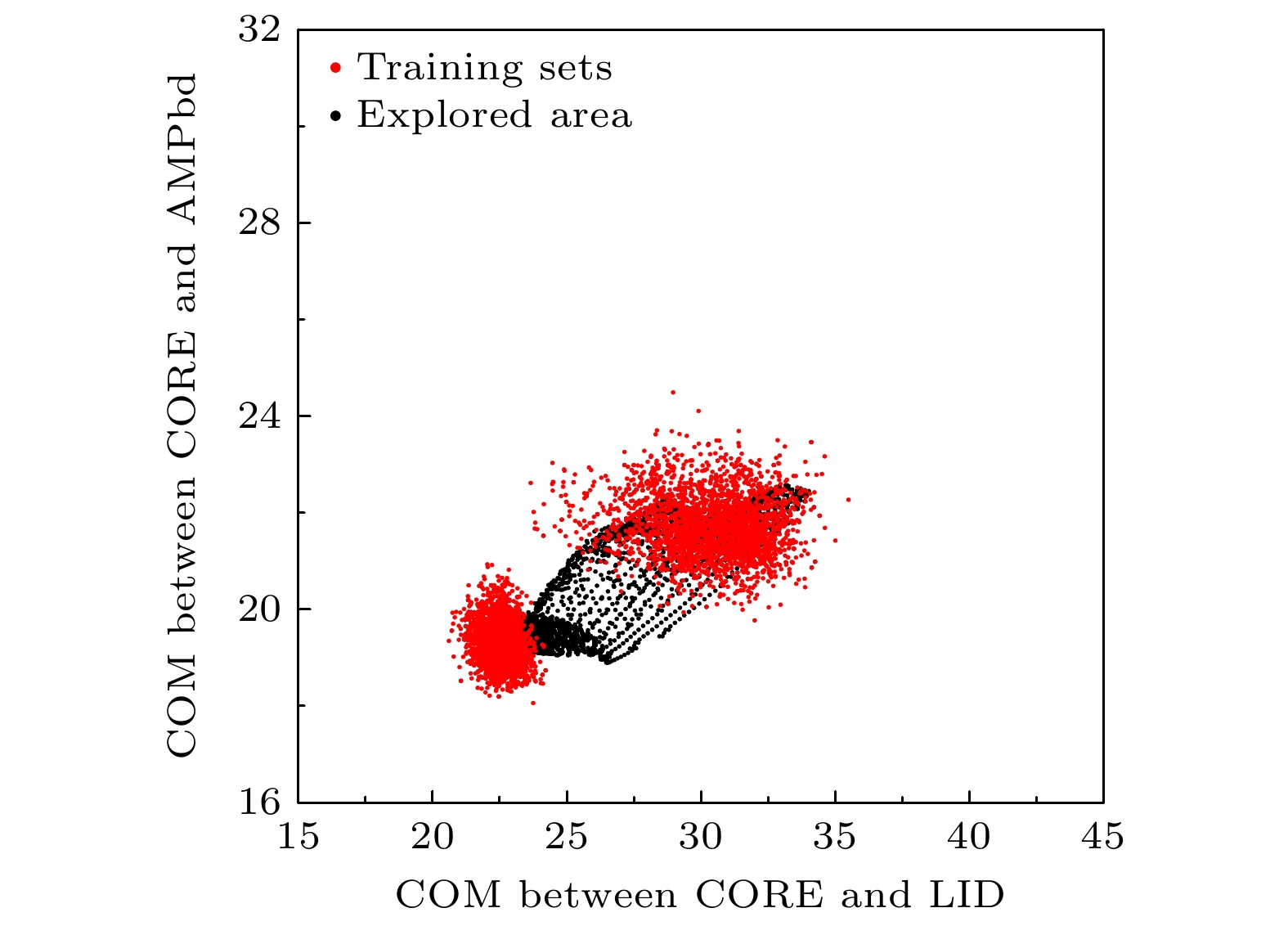
图 3 T4L的构象空间探索结果 (a) 使用AMBER99SB力场/OPC水模型; (b)使用CHARMM36m力场/TIP3P水模型
Fig. 3. Results of conformational space exploration of T4L: (a) With AMBER99SB/OPC; (b) with CHARMM36m/ TIP3P.
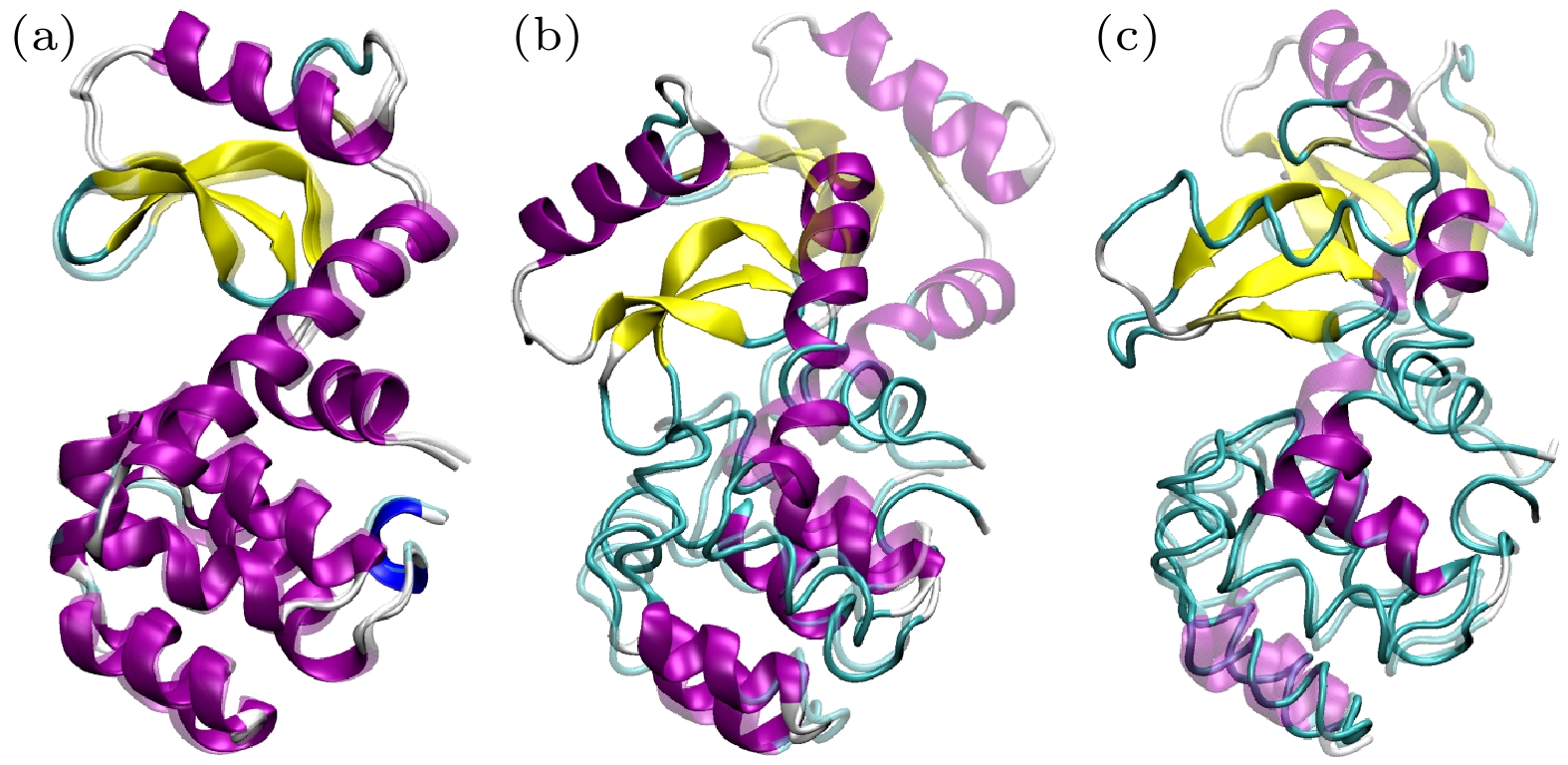
图 4 探索到的不同T4L构象 (a) PDB编号173L的晶体结构(不透明)与探索到的相似结构(透明); (b) 开合程度不同的两个构象; (c) 扭动情况不同的两个构象; 紫色为α螺旋, 黄色为β折叠
Fig. 4. Different T4L conformations explored: (a) PDB:173L (opaque) and a similar structure explored; (b) two conformations with different degrees of opening and closing; (c) two conformations with different degrees of twisting. α-helix is colored in purple and β-sheet is colored in yellow.
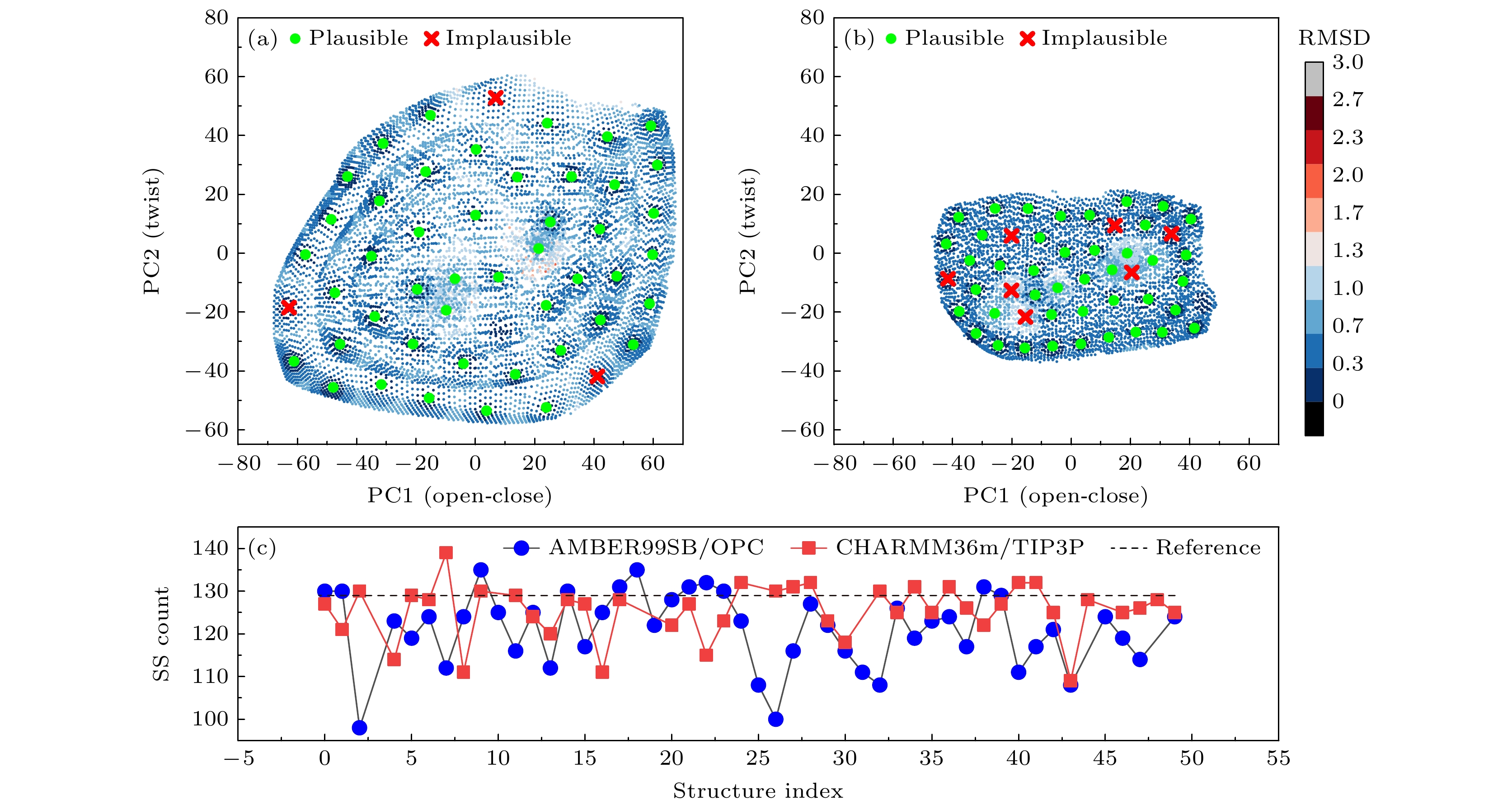
图 5 T4L构象探索结果的合理性检验 (a) 使用AMBER99SB力场/OPC水模型; (b) 使用CHARMM36m力场/TIP3P水模型; (c) 修复后各代表构象的二级结构含量, 参考值为模拟轨迹的平均值
Fig. 5. Plausibility check of T4L conformational exploration results: (a) With AMBER99SB/OPC; (b) with CHARMM36m/TIP3P; (c) secondary structure counts of each representative conformation after fixing, the reference is the average value of the simulated trajectory.
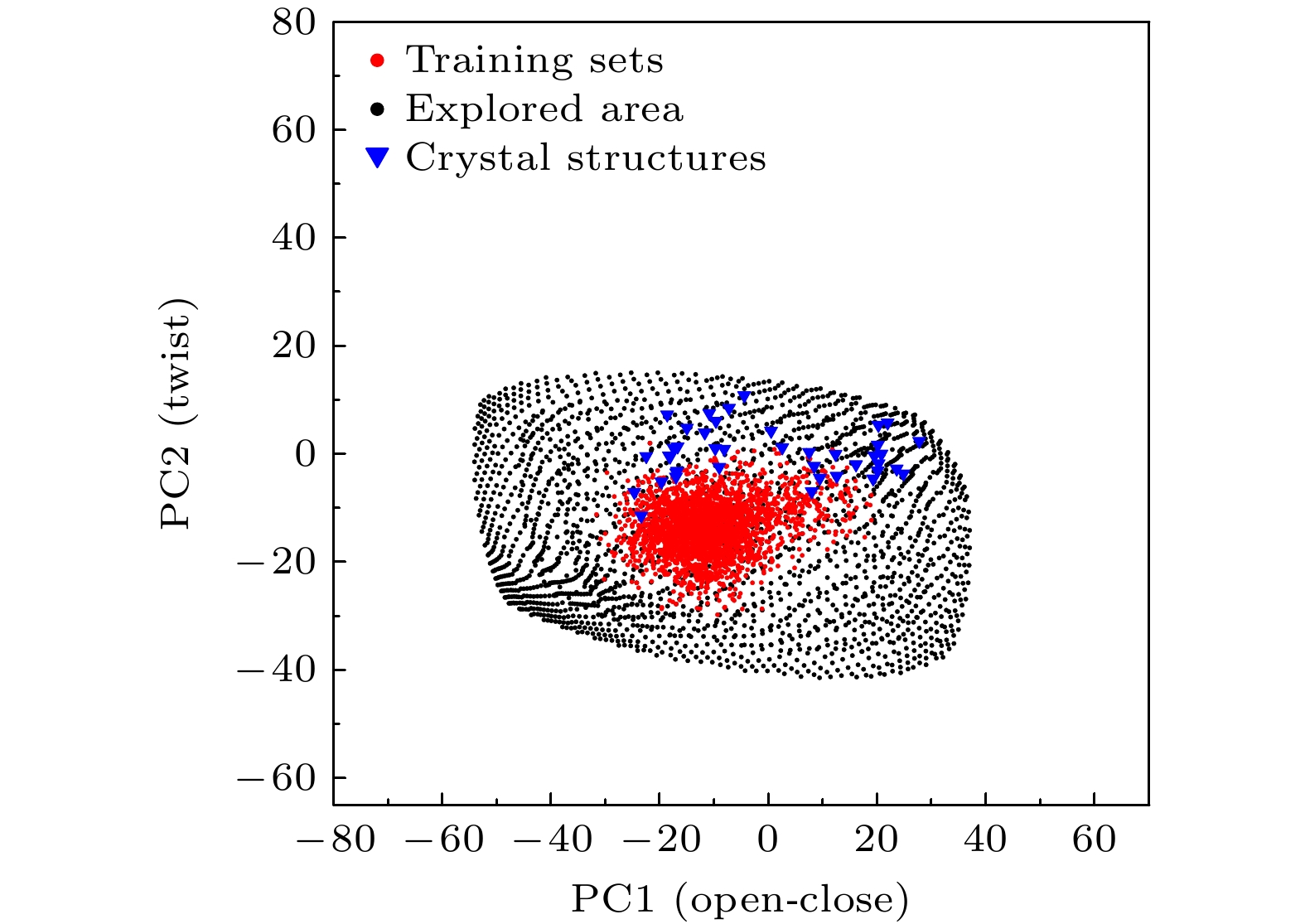
图 6 仅从打开状态出发的T4L构象探索结果
Fig. 6. Results of T4L conformational exploration from the open state only.
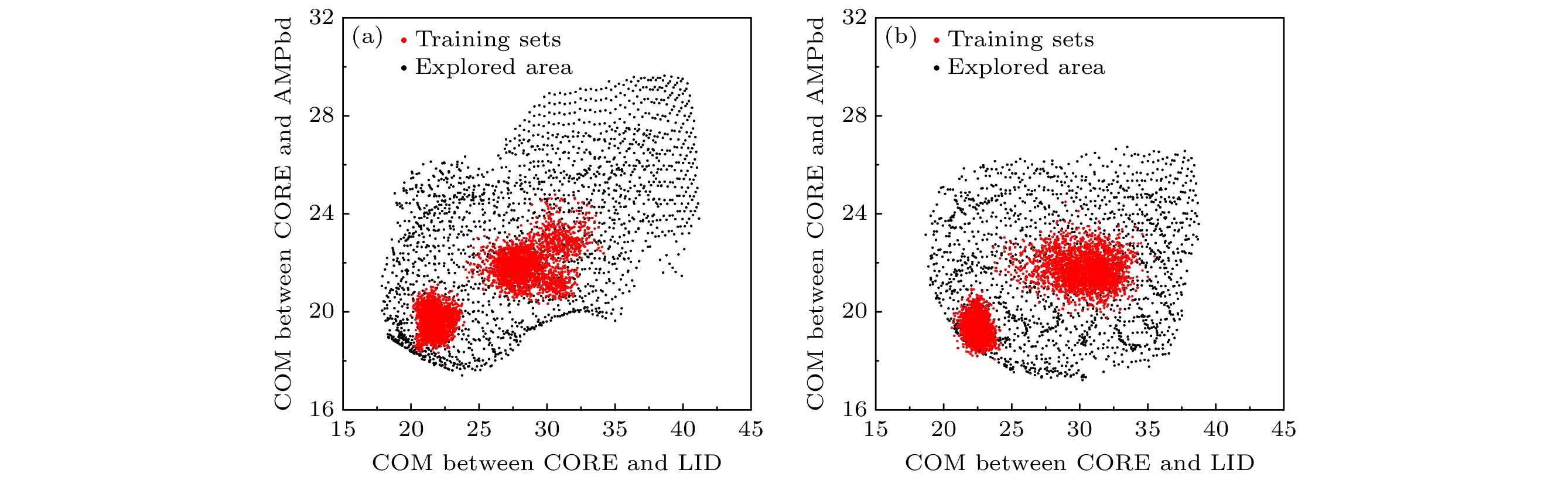
图 7 AdK的构象空间探索结果 (a) 使用AMBER99SB力场/OPC水模型; (b)使用CHARMM36m力场/TIP3P水模型
Fig. 7. Results of conformational space exploration of AdK: (a) With AMBER99SB/OPC; (b) with CHARMM36m/TIP3P.
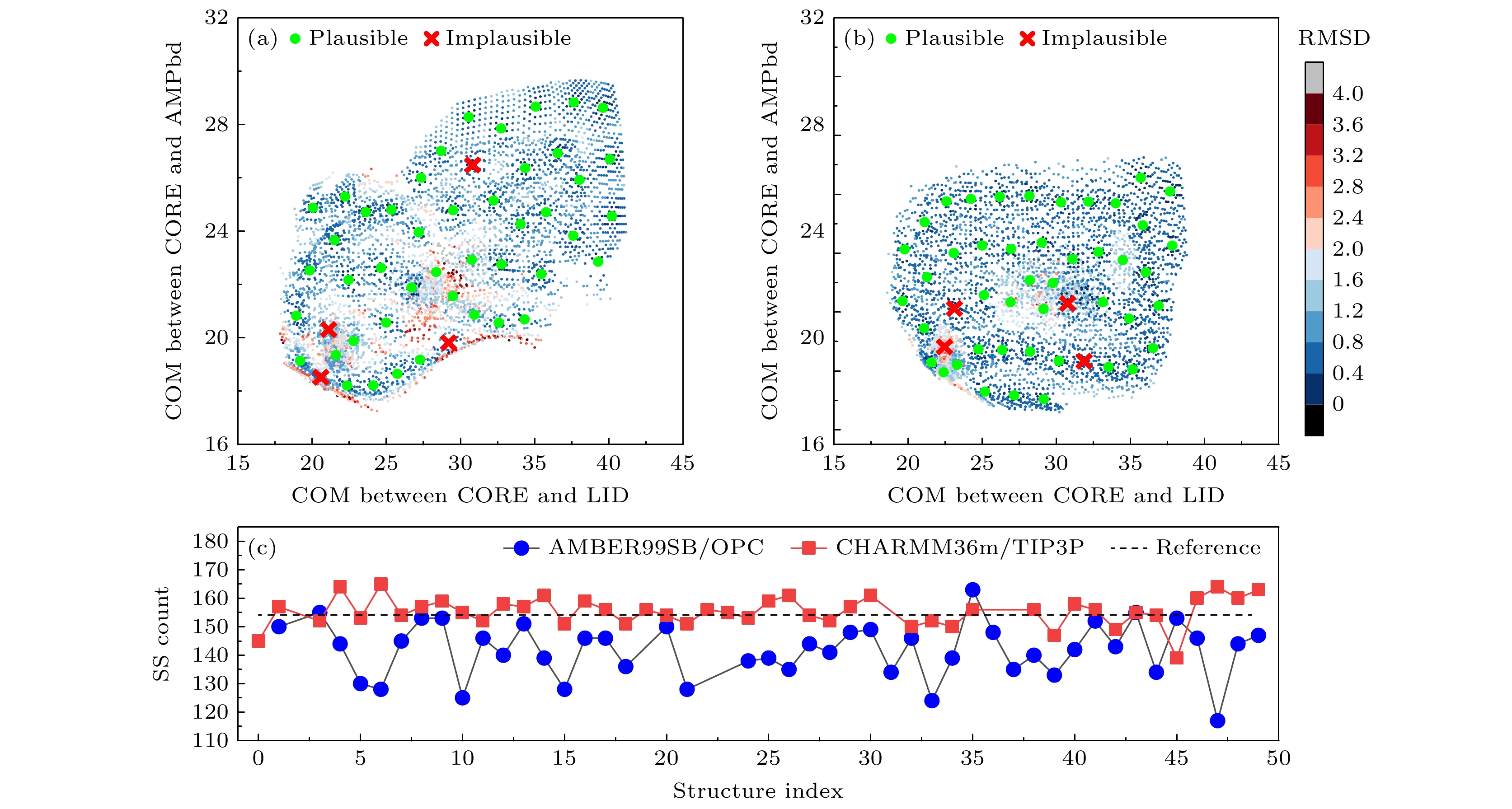
图 9 AdK构象探索结果的合理性检验 (a) 使用AMBER99SB力场/OPC水模型; (b)使用CHARMM36m力场/TIP3P水模型; (c) 修复后各代表构象的二级结构含量, 参考值为模拟轨迹的平均值
Fig. 9. Plausibility check of AdK conformational exploration results: (a) With AMBER99SB/OPC; (b) with CHARMM36m/TIP3P; (c) secondary structure counts of each representative conformation after fixing, the reference is the average value of the simulated trajectory.
-
[1] Chu X, Gan L, Wang E, Wang J 2013 Proc. Natl. Acad. Sci. U.S.A. 110 E2342
Google Scholar
[2] Smyth M S, Martin J H 2000 Mol. Pathol. 53 8
Google Scholar
[3] Danev R, Yanagisawa H, Kikkawa M 2019 Trends Biochem. Sci. 44 837
Google Scholar
[4] Vincenzi M, Mercurio F A, Leone M 2021 Curr. Med. Chem. 28 2729
Google Scholar
[5] Kachala M, Valentini E, Svergun D I 2015 Adv. Exp. Med. Biol. 870 261
Google Scholar
[6] Chu F, Thornton D T, Nguyen H T 2018 Methods 144 53
Google Scholar
[7] Bhaumik S R 2021 Emerg. Top Life Sci. 5 49
Google Scholar
[8] Jumper J, Evans R, Pritzel A, Green T, Figurnov M, Ronneberger O, Tunyasuvunakool K, Bates R, Žídek A, Potapenko A, Bridgland A, Meyer C, Kohl S A A, Ballard A J, Cowie A, Romera-Paredes B, Nikolov S, Jain R, Adler J, Back T, Petersen S, Reiman D, Clancy E, Zielinski M, Steinegger M, Pacholska M, Berghammer T, Bodenstein S, Silver D, Vinyals O, Senior A W, Kavukcuoglu K, Kohli P, Hassabis D 2021 Nature 596 583
Google Scholar
[9] Baek M, DiMaio F, Anishchenko I, Dauparas J, Ovchinnikov S, Lee G R, Wang J, Cong Q, Kinch L N, Schaeffer R D, Millán C, Park H, Adams C, Glassman C R, DeGiovanni A, Pereira J H, Rodrigues A V, van Dijk A A, Ebrecht A C, Opperman D J, Sagmeister T, Buhlheller C, Pavkov-Keller T, Rathinaswamy M K, Dalwadi U, Yip C K, Burke J E, Garcia K C, Grishin N V, Adams P D, Read R J, Baker D 2021 Science 373 871
Google Scholar
[10] Karplus M, Kuriyan J 2005 Proc. Natl. Acad. Sci. 102 6679
Google Scholar
[11] Bernardi R C, Melo M C R, Schulten K 2015 Biochim. Biophys. Acta 1850 872
Google Scholar
[12] Mu J, Liu H, Zhang J, Luo R, Chen H F 2021 J. Chem. Inf. Model. 61 1037
Google Scholar
[13] Lemke T, Peter C 2019 J. Chem. Theory Comput. 15 1209
Google Scholar
[14] Zhu J, Wang J, Han W, Xu D 2022 Nat. Commun. 13 1661
Google Scholar
[15] Hinton G E, Salakhutdinov R R 2006 Science 313 504
Google Scholar
[16] Degiacomi M T 2019 Structure 27 1034
Google Scholar
[17] Wen B, Peng J, Zuo X, Gong Q, Zhang Z 2014 Biophysical J. 107 956
Google Scholar
[18] Giri Rao V V H, Gosavi S 2014 PLOS Computational Biology 10 e1003938
Google Scholar
[19] Abraham M J, Murtola T, Schulz R, Páll S, Smith J C, Hess B, Lindahl E 2015 SoftwareX 1–2 19
Google Scholar
[20] Weaver L H, Matthews B W 1987 J. Mol. Biol. 193 189
Google Scholar
[21] Zhang X J, Wozniak J A, Matthews B W 1995 J. Mol. Biol. 250 527
Google Scholar
[22] Müller C W, Schulz G E 1992 J. Mol. Biol. 224 159
Google Scholar
[23] Müller C W, Schlauderer G J, Reinstein J, Schulz G E 1996 Structure 4 147
Google Scholar
[24] Hornak V, Abel R, Okur A, Strockbine B, Roitberg A, Simmerling C 2006 Proteins Struct. Funct. Bioinf. 65 712
Google Scholar
[25] Izadi S, Anandakrishnan R, Onufriev A V 2014 J. Phys. Chem. Lett. 5 3863
Google Scholar
[26] Huang J, Rauscher S, Nawrocki G, Ran T, Feig M, de Groot B L, Grubmüller H, MacKerell A D 2017 Nat. Methods 14 71
Google Scholar
[27] Bussi G, Donadio D, Parrinello M 2007 J. Chem. Phys. 126 014101
Google Scholar
[28] Essmann U, Perera L E, Berkowitz M L, Darden T A, Lee H C, Pedersen L G 1995 J. Chem. Phys. 103 8577
Google Scholar
[29] Kingma D P, Ba J 2014 arXiv:1412.6980 [cs.LG
[30] Lovell S C, Davis I W, Arendall III W B, de Bakker P I W, Word J M, Prisant M G, Richardson J S, Richardson D C 2003 Proteins Struct. Funct. Bioinf. 50 437
Google Scholar
[31] Eastman P, Swails J, Chodera J D, McGibbon R T, Zhao Y, Beauchamp K A, Wang L P, Simmonett A C, Harrigan M P, Stern C D, Wiewiora R P, Brooks B R, Pande V S 2017 PLoS Comput. Biol. 13 e1005659
Google Scholar
[32] Shirts M R, Klein C, Swails J M, Yin J, Gilson M K, Mobley D L, Case D A, Zhong E D 2017 J. Comput. -Aided Mol. Des. 31 147
Google Scholar
[33] Touw W G, Baakman C, Black J, te Beek T A, Krieger E, Joosten R P, Vriend G 2015 Nucleic Acids Res. 43 D364
Google Scholar

 下载:
下载:
计量
- 文章访问数: 6181
- PDF下载量: 209
- 被引次数: 0
