生物分子模拟中的机器学习
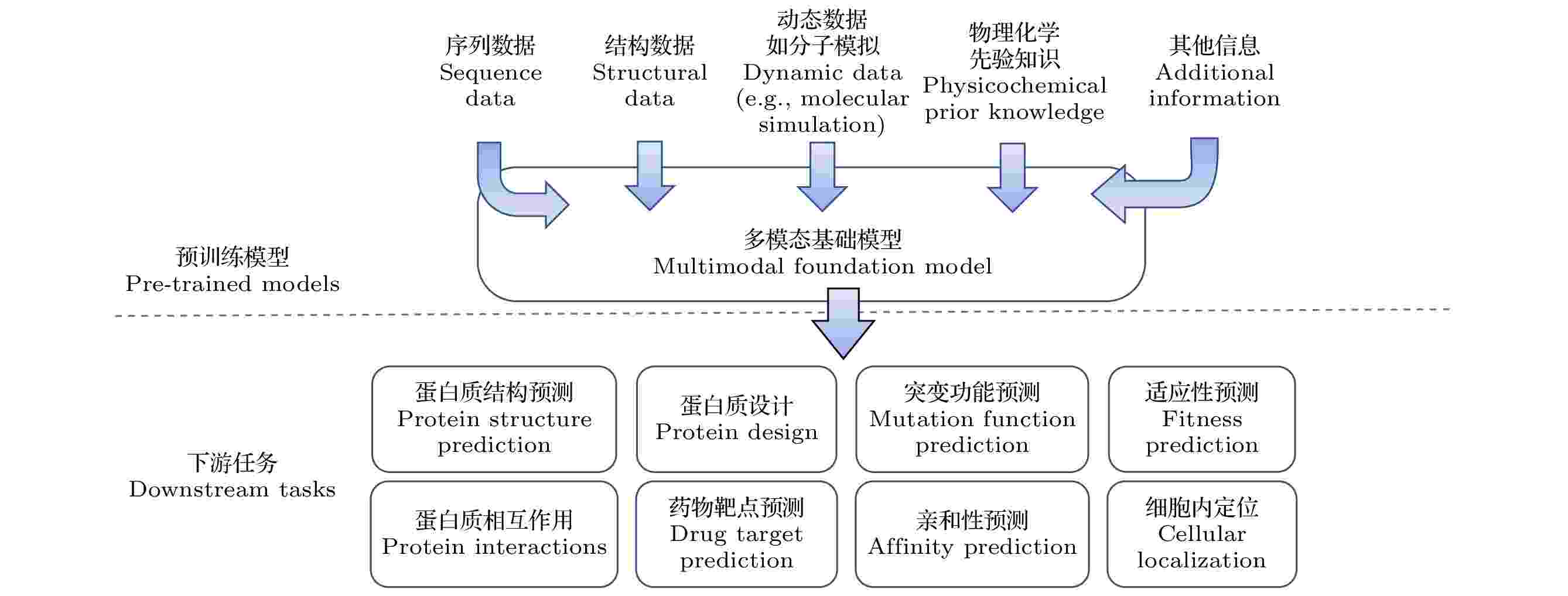
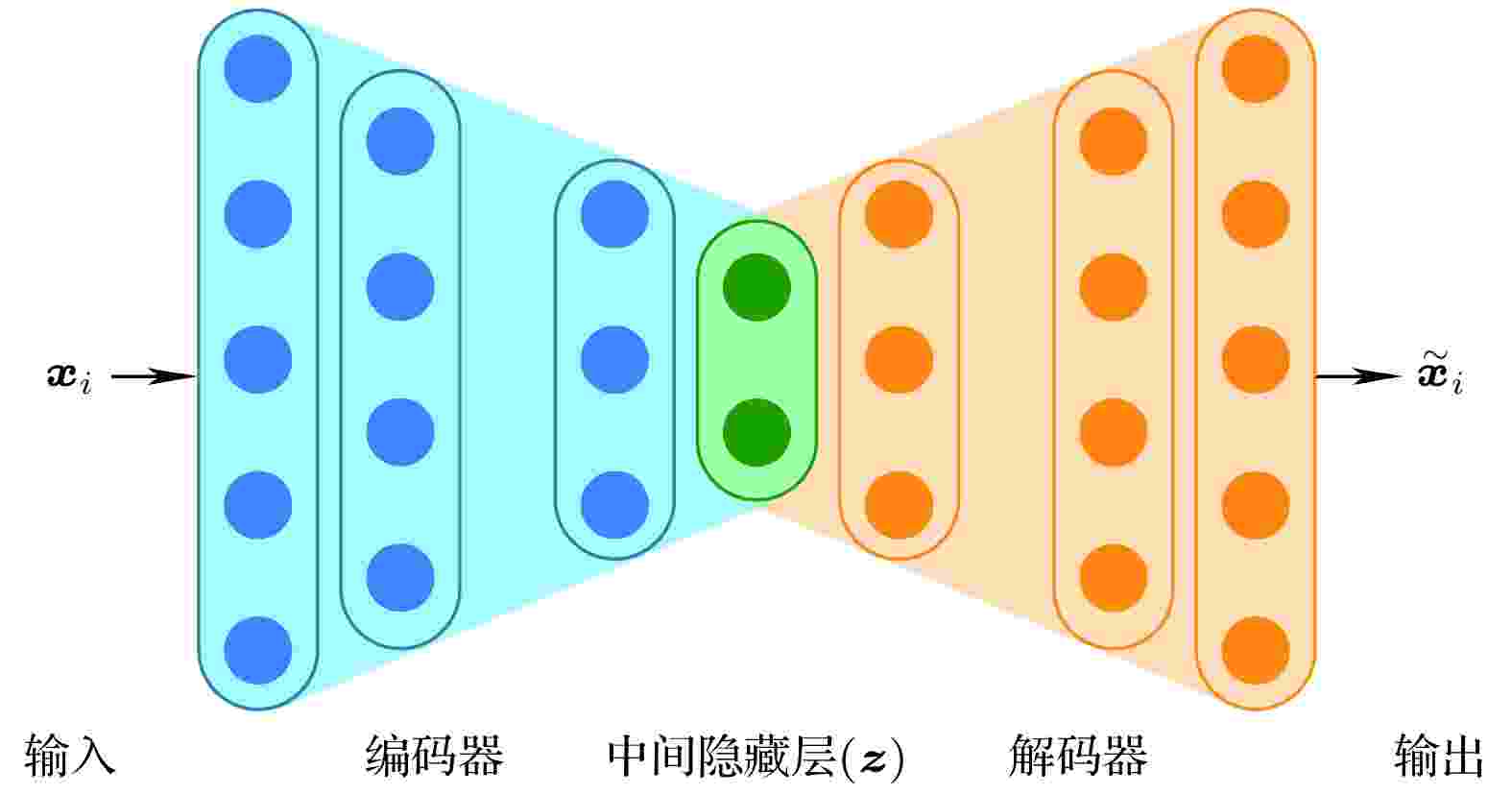
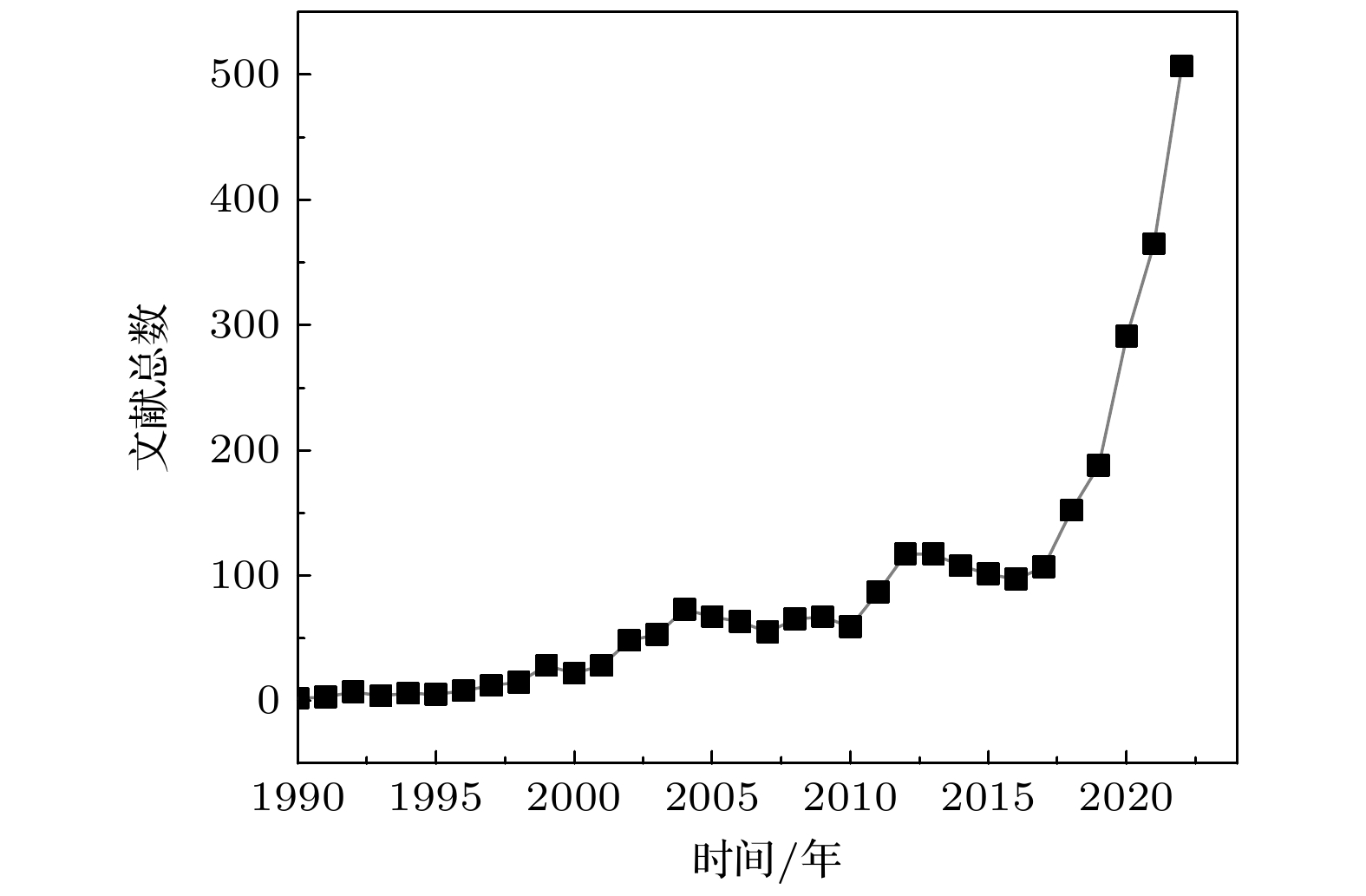
分子模拟技术是人们从分子层次探究生命现象物理原理的重要手段, 被广泛应用于蛋白质等生物大分子的结构与动力学研究. 自从 20世纪 70年代 Karplus 等科学家首次将分子动力学模拟应用于蛋白质研究以来, 分子模拟技术在生物分子体系研究中的应用范围不断扩展, 深刻影响了生物物理学与分子生物学研究的基本范式. 生物大分子的结构动力学涉及皮秒到毫秒甚至更长时间尺度,如何精确表征具有复杂能量面特征的生物大分子结构与动力学的多尺度特性是生物分子模拟领域的核心难题. 通过物理、化学以及计算机科学等多个领域科学家近 50年的不懈努力, 人们在生物分子力场准确度提升、各种相互作用的准确描述和计算、增强采样与自由能计算、高维分子模拟数据信息挖掘以及多尺度理论模拟算法构建等方面取得了多个突破. 目前, 人们不仅能够实现对一些蛋白质分子体系毫秒时间尺度的折叠全过程进行分子模拟, 而且能够实现对病毒颗粒、细胞质、甚至染色质等超大分子体系进行分子模拟, 在推动生命科学研究向定量化转变中发挥了重要作用. 近年来, 机器学习技术的突飞猛进为解决生物分子模拟中的挑战难题提供了新思路. 人们开始广泛利用深度学习技术构建高精度分子力场、增强分子模拟采样效率、分析高维复杂的分子模拟数据、提取结构及动力学特征等, 取得了一系列重要进展. 结合机器学习算法的分子模拟技术已经在生物物理机制探究、药物设计、结构与动力学预测等基础与应用研究中展现出其实用性与巨大发展潜力.
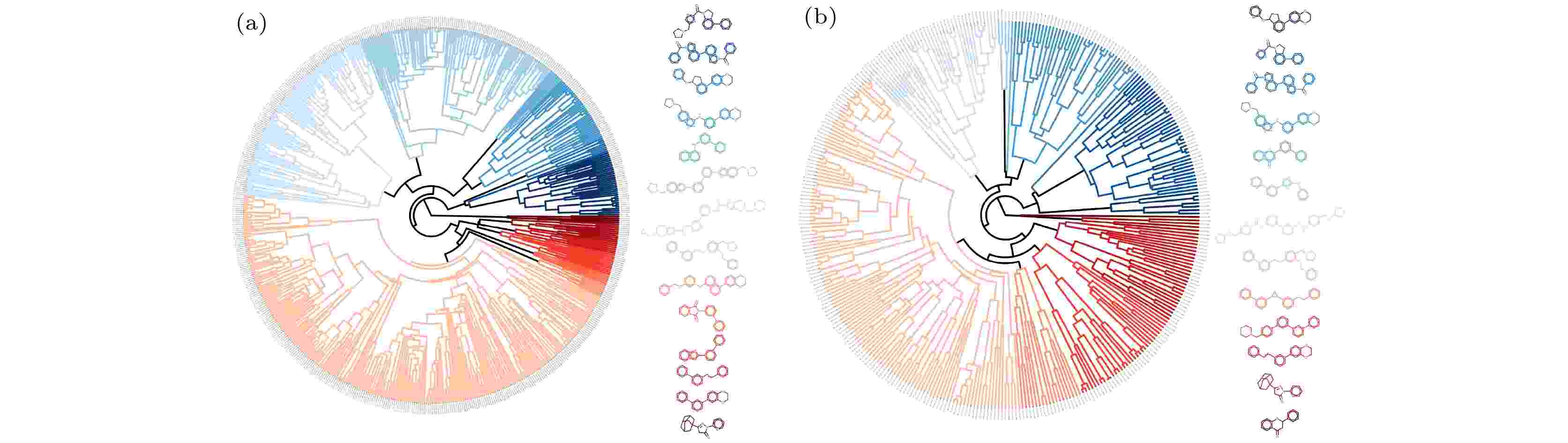
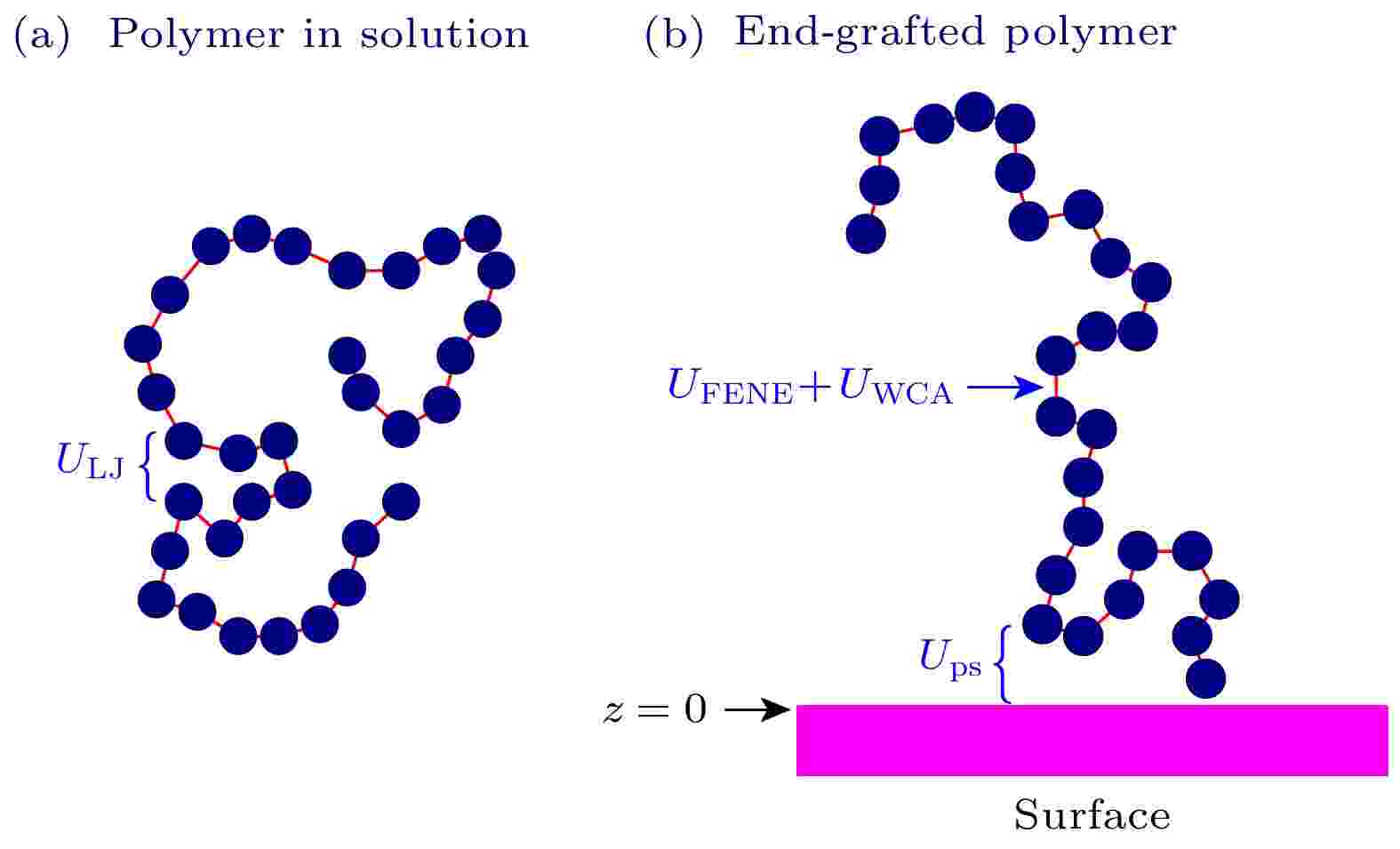
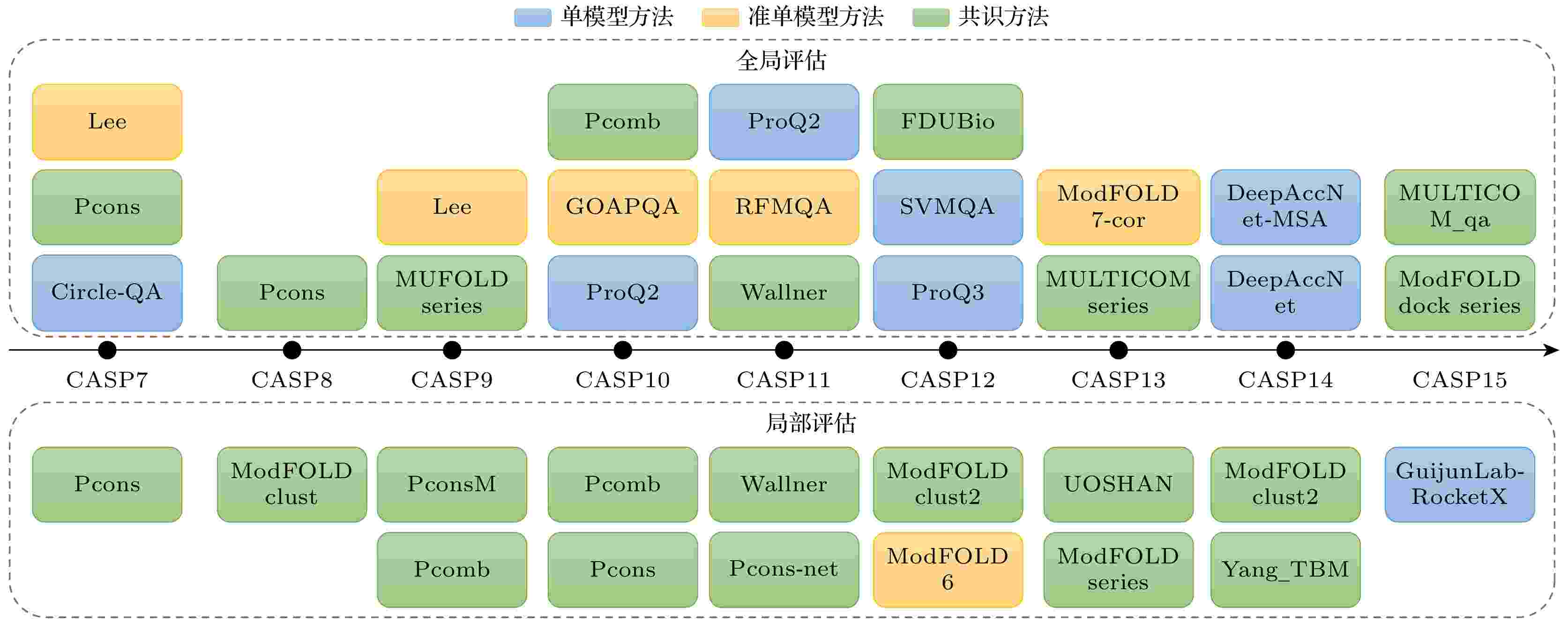
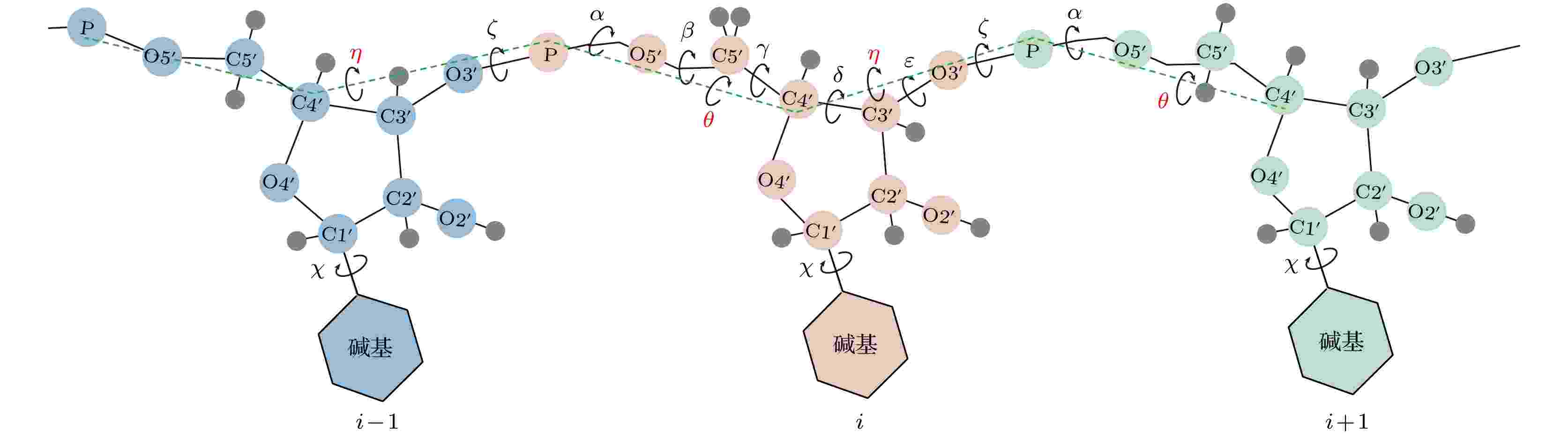
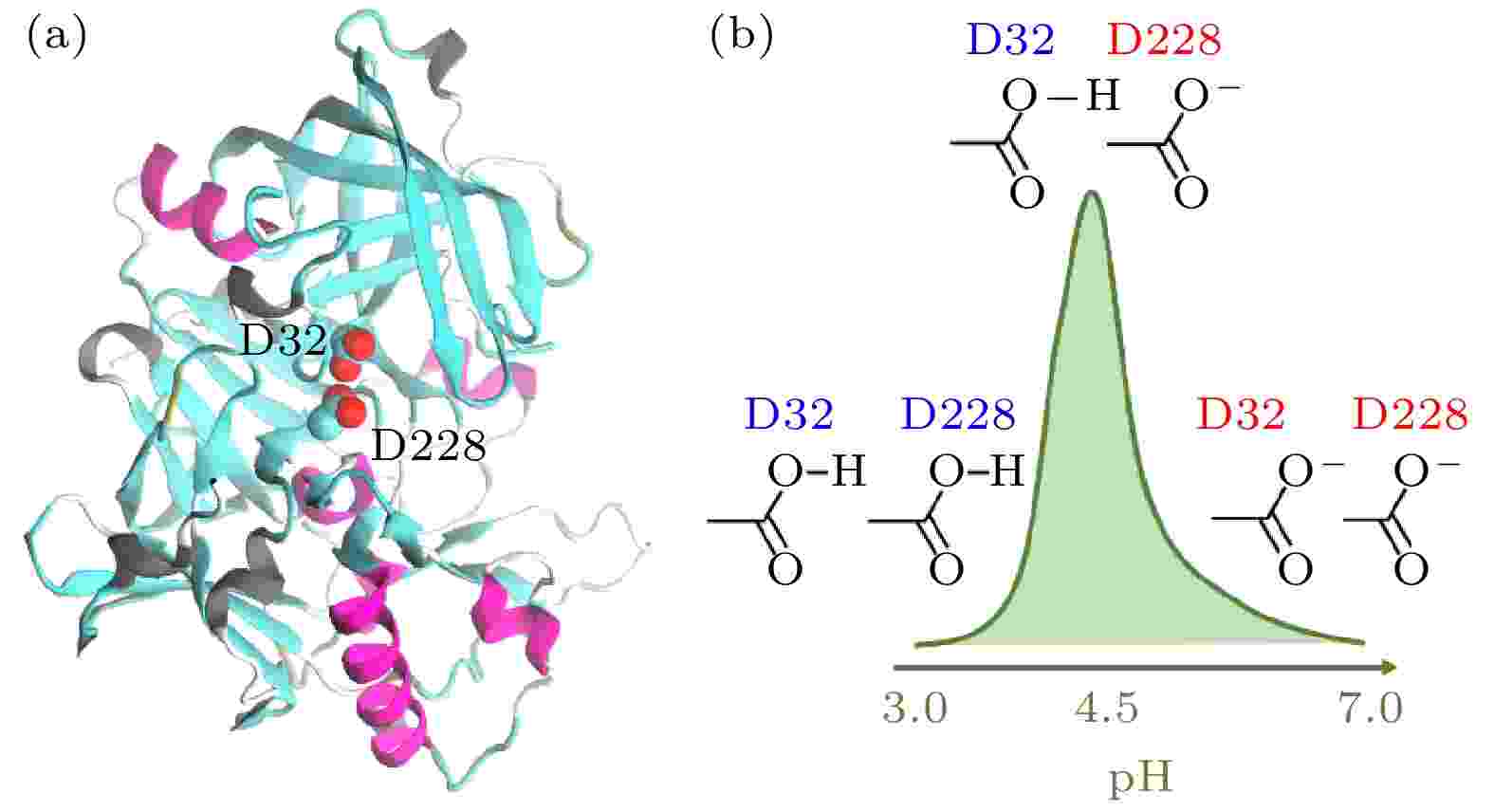
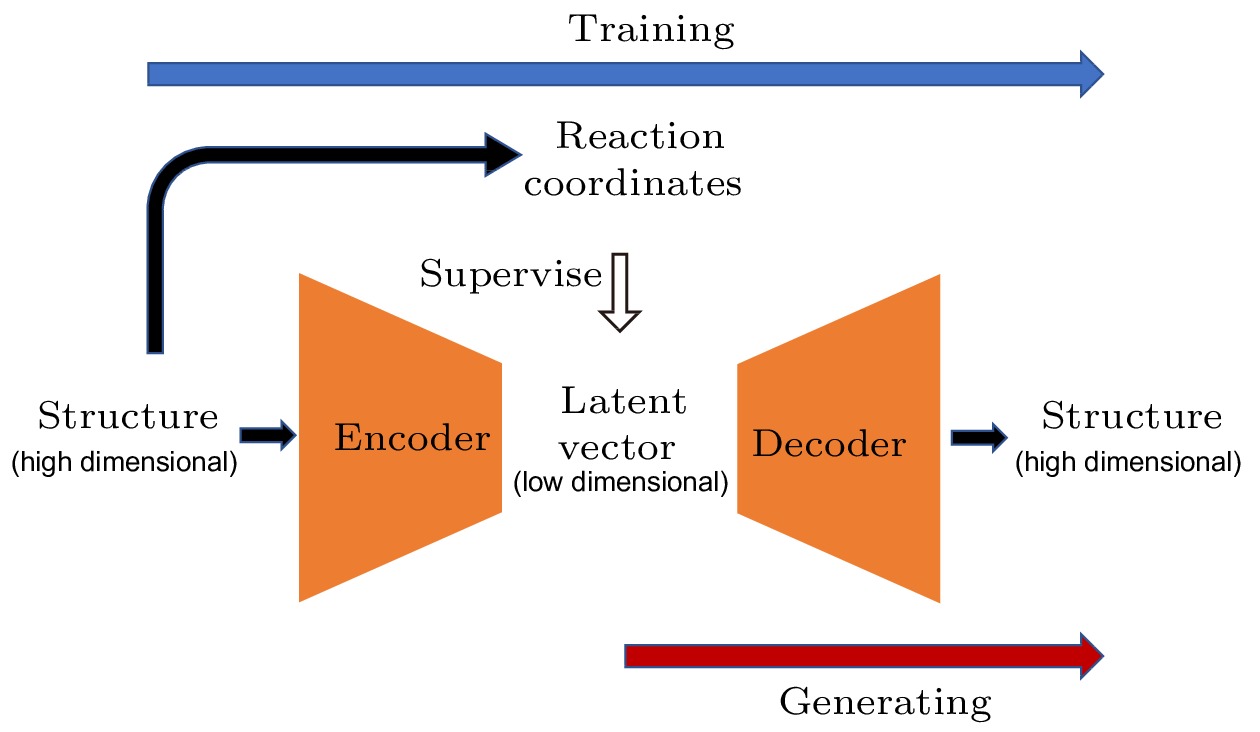
鉴于机器学习算法在推动生物分子模拟技术发展和生物物理研究中的关键作用, 《物理学报》特组织本专题, 邀请国内部分活跃在该领域前沿的学者撰稿, 深入探讨生物分子模拟与机器学习融合应用的最新研究成果, 并对该领域当前面临的重要挑战及未来研究中可能的突破方向进行综述和展望. 相关论文涵盖了基于机器学习算法的蛋白质分子模拟构象空间搜索、RNA扭转角预测、蛋白质等生物大分子 pKa值预测、生物大分子构象过渡态搜索、蛋白质结构模型质量评估、靶标特异性药物筛选、蛋白质分子设计、高分子塌缩相变和临界吸附相变以及分子体系高维自由能地貌图构建等十余篇研究和综述论文, 分两期刊出. 这些研究论文和综述从不同的角度展示了国内外该领域的最新进展和研究现状. 希望本专题有助于读者了解该领域的前沿研究课题, 并能对促进国内生物分子模拟学术交流发挥作用. 本专题讨论的研究领域涉及多个学科的交叉融合, 且突破性的研究成果不断涌现, 因此本专题所涵盖的代表性成果和前沿进展介绍难免有所遗漏, 不足之处敬请谅解.