










-
蛋白质计算一直以来都是科学领域中的重要课题, 而近年来其与机器学习的结合, 更是极大地推进了相关学科的发展. 本综述主要讨论了机器学习在四个重要的蛋白质计算领域内的研究进展, 这四个领域包括:分子动力学模拟、结构预测、性质预测和分子设计. 分子动力学模拟依赖于力场参数, 准确的力场参数是分子动力学模拟的必需品, 而机器学习可以帮助研究者得到更加准确的力场参数. 在分子动力学模拟中, 机器学习也可以从复杂的体系中以较小的代价计算出所需求解的自由能. 结构预测一般是给定蛋白质序列预测其结构. 结构预测复杂度高、数据量大, 而这恰恰是机器学习所擅长的. 在机器学习的协助下, 近年来科研人员已经在单个蛋白质三维结构预测上取得了不错的成果. 性质预测则是指通过给定的已知蛋白质信息, 推断其可能拥有的性质, 这对于蛋白质的研究也是至关重要的. 更具挑战性的是分子设计, 虽然近年来机器学习在蛋白质设计上取得突破, 但这一领域还有很大空间值得探索. 本综述将针对以上四点分别展开论述, 并对蛋白质计算中的机器学习研究进行展望.In silico protein calculation has been an important research subject for a long time, while its recent combination with machine learning promotes the development greatly in related areas. This review focuses on four major fields of the in silico protein research that combines with machine learning, which are molecular dynamics, structure prediction, property prediction and molecule design. Molecular dynamics depend on the parameters of force field, which is necessary for obtaining accurate results. Machine learning can help researchers to obtain more accurate force field parameters. In molecular dynamics simulation, machine learning can also help to perform the free energy calculation in relatively low cost. Structure prediction is generally used to predict the structure given a protein sequence. Structure prediction is of high complexity and data volume, which is exactly what machine learning is good at. By the help of machine learning, scientists have gained great achievements in three-dimensional structure prediction of proteins. On the other hand, the predicting of protein properties based on its known information is also important to study protein. More challenging, however, is molecule design. Though marching learning has made breakthroughs in drug-like small molecule design and protein design in recent years, there is still plenty of room for exploration. This review focuses on summarizing the above four fields andlooks forward to the application of marching learning to the in silico protein research.
-
Keywords:
- protein /
- machine learning /
- molecular dynamics simulation /
- structural prediction /
- properties prediction /
- molecular design
[1] Baltoumas F A, Zafeiropoulou S, Karatzas E, et al. 2021 Biomolecules 11 1245
 Google Scholar
Google Scholar
[2] Wolf Y I, Katsnelson M I, Koonin E V 2018 Proc. Natl. Acad. Sci. USA 115 E8678
Google Scholar
[3] Fusco A, Fedele M 2007 Nat. Rev. Cancer 7 899
Google Scholar
[4] Noble D 2002 Nat. Rev. Mol. Cell Biol. 3 459
Google Scholar
[5] Markowetz F 2017 PLoS Biology 15 e2002050
Google Scholar
[6] Hollingsworth S A, Dror R O 2018 Neuron 99 1129
Google Scholar
[7] Zhang Y 2008 Curr. Opin. Struct. Biol. 18 342
Google Scholar
[8] Agostini F, Vendruscolo M, Tartaglia G G 2012 J. Mol. Biol. 421 237
Google Scholar
[9] Chen L, Fan Z, Chang J, et al. 2023 Nat. Commun. 14 4217
Google Scholar
[10] Geng H, Chen F, Ye J, Jiang F 2019 Computat. Struct. Biotechnol. J. 17 1162
Google Scholar
[11] Salo-Ahen O M, Alanko I, Bhadane R, et al. 2020 Processes 9 71
Google Scholar
[12] Norberg J, Nilsson L 2003 Q. Rev. Biophys. 36 257
Google Scholar
[13] van der Kamp M W, Shaw K E, Woods C J, Mulholland A J 2008 J. R. Soc. Interface 5 173
Google Scholar
[14] Dror R O, Dirks R M, Grossman J, Xu H, Shaw D E 2012 Annu. Rev. Biophys. 41 429
Google Scholar
[15] Lin X, Li X, Lin X 2020 Molecules 25 1375
Google Scholar
[16] Pearce R, Zhang Y 2021 Curr. Opin. Struct. Biol. 68 194
Google Scholar
[17] Jordan M I, Mitchell T M 2015 Science 349 255
Google Scholar
[18] Butler K T, Davies D W, Cartwright H, Isayev O, Walsh A 2018 Nature 559 547
Google Scholar
[19] Liakos K G, Busato P, Moshou D, Pearson S, Bochtis D 2018 Sensors 18 2674
Google Scholar
[20] Jiang T, Gradus J L, Rosellini A J 2020 Behav. Ther. 51 675
Google Scholar
[21] Hastie T, Tibshirani R, Friedman J, Hastie T, Tibshirani R, Friedman J 2009 Unsupervised Learning. In: The Elements of Statistical Learning. Springer Series in Statistics (New York: Springer) pp485–585
[22] Van Engelen J E, Hoos H H 2020 Machine Learning 109 373
Google Scholar
[23] Wiering M A, Van Otterlo M 2012 Reinforcement Learning (Heidelberg, Berlin: Springer) p729
[24] LeCun Y, Bengio Y, Hinton G 2015 Nature 521 436
Google Scholar
[25] Deng L, Yu D 2014 Deep Learning: Methods and Applications (Now Foundations and Trends) p197
[26] Jones D T 2019 Nat. Rev. Mol. Cell Biol. 20 659
Google Scholar
[27] Das P, Sercu T, Wadhawan K, et al. 2021 Nat. Biomed. Eng. 5 613
Google Scholar
[28] Kuhlman B, Bradley P 2019 Nat. Rev. Mol. Cell Biol. 20 681
Google Scholar
[29] Trevino S R, Scholtz J M, Pace C N 2008 J. Pharm. Sci. 97 4155
Google Scholar
[30] Kelley K W, Weigent D A, Kooijman R 2007 Brain Behav. Immun. 21 384
Google Scholar
[31] Babin V, Roland C, Sagui C 2008 J. Chem. Phys. 128
Google Scholar
[32] Morozov I V, Kazennov A M, Bystryi R, Norman G E, Pisarev V V, Stegailov V V 2011 Comput. Phys. Commun. 182 1974
Google Scholar
[33] Karplus M, McCammon J A 2002 Nat. Struct. Biol. 9 646
Google Scholar
[34] Wang Y, Ribeiro J M L, Tiwary P 2020 Curr. Opin. Struct. Biol. 61 139
Google Scholar
[35] Chmiela S, Tkatchenko A, Sauceda H E, Poltavsky I, Schütt K T, Müller K R 2017 Sci. Adv. 3 e1603015
Google Scholar
[36] Ponder J W, Case D A 2003 Adv. Protein Chem. 66 27
Google Scholar
[37] Monticelli L, Tieleman D P 2013 Biomolecular Simulations: Methods and Protocols 197
[38] Wang J, Wolf R M, Caldwell J W, Kollman P A, Case D A 2004 J. Comput. Chem. 25 1157
Google Scholar
[39] Hughes Z E, Wright L B, Walsh T R 2013 Langmuir 29 13217
Google Scholar
[40] Cesari A, Bottaro S, Lindorff-Larsen K, Banáš P, Šponer J, Bussi G 2019 J. Chem. Theory Comput. 15 3425
Google Scholar
[41] Unke O T, Chmiela S, Sauceda H E, Gastegger M, Poltavsky I, Schütt K T, Tkatchenko A, Müller K R 2021 Chem. Rev. 121 10142
Google Scholar
[42] Poltavsky I, Tkatchenko A 2021 J. Phys. Chem. Lett. 12 6551
Google Scholar
[43] Kästner J 2011 WIREs Comput. Mol. Sci. 1 932
Google Scholar
[44] Izrailev S, Stepaniants S, Isralewitz B, Kosztin D, Lu H, Molnar F, Wriggers W, Schulten K 1999 Computational Molecular Dynamics: Challenges, Methods, Ideas: Proceedings of the 2nd International Symposium on Algorithms for Macromolecular Modelling Berlin, May 21–24, 1997 p39
[45] Moradi M, Babin V, Roland C, Sagui C 2013 Nucleic Acids Res. 41 33
Google Scholar
[46] Simonson T, Archontis G, Karplus M 2002 Acc. Chem. Res. 35 430
Google Scholar
[47] Bitencourt-Ferreira G, de Azevedo W F 2018 Biophys. Chem. 240 63
Google Scholar
[48] Trott O, Olson A J 2010 J. Comput. Chem. 31 455
Google Scholar
[49] Besora M, Vidossich P, Lledos A, Ujaque G, Maseras F 2018 J. Phys. Chem. A 122 1392
Google Scholar
[50] Pan X, Yang J, Van R, Epifanovsky E, Ho J, Huang J, Pu J, Mei Y, Nam K, Shao Y 2021 J. Chem. Theory Comput. 17 5745
Google Scholar
[51] Senn H M, Thiel W 2009 Angew. Chem. Int. Ed. 48 1198
Google Scholar
[52] Riniker S 2017 J. Chem. Inf. Model. 57 726
Google Scholar
[53] Bennett W D, He S, Bilodeau C L, Jones D, Sun D, Kim H, Allen J E, Lightstone F C, Ingólfsson H I 2020 J. Chem. Inf. Model. 60 5375
Google Scholar
[54] Bertazzo M, Gobbo D, Decherchi S, Cavalli A 2021 J. Chem. Theory Comput. 17 5287
Google Scholar
[55] Eswar N, John B, Mirkovic N, et al. 2003 Nucleic Acids Research 31 3375
Google Scholar
[56] Asara J M, Schweitzer M H, Freimark L M, Phillips M, Cantley L C 2007 Science 316 280
Google Scholar
[57] Greener J G, Kandathil S M, Moffat L, Jones D T 2022 Nat. Rev. Mol. Cell Biol. 23 40
Google Scholar
[58] Jumper J, Evans R, Pritzel A, et al. 2021 Nature 596 583
Google Scholar
[59] Wu R, Ding F, Wang R, et al. 2022 bioRxiv 2022.07.21. 500999
[60] Baek M, DiMaio F, Anishchenko I, et al. 2021 Science 373 871
Google Scholar
[61] Medsker L R, Jain L 1999 Recurrent Neural Networks: Design and Applications (1st Ed.) (CRC Press) p2
[62] Kim P 2017 Convolutional Neural Network. In: MATLAB Deep Learning (Berkeley, CA: Apress) p121
[63] Wardah W, Khan M G, Sharma A, Rashid M A 2019 Comput. Biol. Chem. 81 1
Google Scholar
[64] Mirabello C, Pollastri G 2013 Bioinformatics 29 2056
Google Scholar
[65] Heffernan R, Yang Y, Paliwal K, Zhou Y 2017 Bioinformatics 33 2842
Google Scholar
[66] Wang S, Peng J, Ma J, Xu J 2016 Sci. Rep. 6 1
Google Scholar
[67] Li Z, Yu Y 2016 arXiv: 1604.07176 [q-bio.BM]
[68] Wang Y, Mao H, Yi Z 2017 Knowledge-Based Systems 118 115
Google Scholar
[69] Nishikawa K, Ooi T, Isogai Y, Saitô N 1972 J. Phys. Soc. JPN 32 1331
Google Scholar
[70] Edgar R C, Batzoglou S 2006 Curr. Opin. Struct. Biol. 16 368
Google Scholar
[71] Vaswani A, Shazeer N, Parmar N, Uszkoreit J, Jones L, Gomez A N, Kaiser Ł, Polosukhin I 2017 Advances in Neural Information Processing Systems 30 Long Beach, USA, December 4–9, 2017 p30
[72] Janin J, Bahadur R P, Chakrabarti P 2008 Q. Rev. Biophys. 41 133
Google Scholar
[73] Zafferani M, Hargrove A E 2021 Cell Chem. Biol. 28 594
Google Scholar
[74] Hunter C A 2004 Angew. Chem. Int. Ed. 43 5310
Google Scholar
[75] Chen R, Li L, Weng Z 2003 Proteins Struct. Funct. Bioinf. 52 80
Google Scholar
[76] Jingcheng Y, Zhaoming C, Zhaoqun L, Mingliang Z, Wenjun L, He H, Qiwei Y 2022 Code of Open Complex https:// github.com/baaihealth/OpenComplex.
[77] Evans R, O’ Neill M, Pritzel A, et al. 2021 bioRxiv 2021.10.04.463034
[78] Moriwaki Y 2021 Twitter https://twitter.com/Ag_smith/ status.
[79] Ko J, Lee J 2021 bioRxiv 2021.07.27.453972 Ko J, Lee J 2021 bioRxiv 2021.07.27.453972
[80] Tsaban T, Varga J K, Avraham O, Ben-Aharon Z, Khramushin A, Schueler-Furman O 2022 Nat. Commun. 13 176
Google Scholar
[81] Bryant P, Pozzati G, Elofsson A 2022 Nat. Commun. 13 1265
Google Scholar
[82] Zhou T M, Wang S, Xu J 2017 bioRxiv 240754
[83] Cang Z, Wei G W 2017 PLoS Comput. Biol. 13 e1005690
Google Scholar
[84] Yagi K, Re S, Mori T, Sugita Y 2022 Curr. Opin. Struct. Biol. 72 88
Google Scholar
[85] Vendruscolo M, Knowles T P, Dobson C M 2011 CSH Perspect. Biol. 3 a010454
Google Scholar
[86] Khurana S, Rawi R, Kunji K, Chuang G Y, Bensmail H, Mall R 2018 Bioinformatics 34 2605
Google Scholar
[87] Wu X, Yu L 2021 Bioinformatics 37 4314
Google Scholar
[88] Schellekens H 2003 Nephrology Dialysis Transplantation 18 1257
Google Scholar
[89] Ternette N, Tippler B, Überla K, Grunwald T 2007 Vaccine 25 7271
Google Scholar
[90] Jefferis R 2016 J. Immunol. Res. 2016
Google Scholar
[91] Schellekens H 2005 Nephrology Dialysis Transplantation 20 vi3
Google Scholar
[92] Smith C C, Chai S, Washington A R, et al. 2019 Cancer Immunol. Res. 7 1591
Google Scholar
[93] Gonzalez-Dias P, Lee E K, Sorgi S, de Lima D S, Urbanski A H, Silveira E L, Nakaya H I 2020 Hum. Vacc. Immunother. 16 269
Google Scholar
[94] Timr S, Madern D, Sterpone F 2020 Prog. Mol. Biol. Transl. Sci. 170 239
Google Scholar
[95] Pudžiuvelytė I, Olechnovič K, Godliauskaite E, Sermokas K, Urbaitis T, Gasiunas G, Kazlauskas D 2023 bioRxiv 2023.03.27.534365 Pudžiuvelytė I, Olechnovič K, Godliauskaite E, Sermokas K, Urbaitis T, Gasiunas G, Kazlauskas D 2023 bioRxiv 2023.03.27.534365
[96] Rives A, Meier J, Sercu T, et al. 2021 Proc. Natl. Acad. Sci. U.S.A. 118 e2016239118
Google Scholar
[97] Elnaggar A, Heinzinger M, Dallago C, et al. 2022 IEEE Trans. Pattern Anal. Mach. Intell. 44 7112
Google Scholar
[98] Huang P S, Boyken S E, Baker D 2016 Nature 537 320
Google Scholar
[99] Huang B, Xu Y, Hu X, Liu Y, Liao S, Zhang J, Huang C, Hong J, Chen Q, Liu H 2022 Nature 602 523
Google Scholar
[100] Watson J L, Juergens D, Bennett N R, et al. 2023 Nature 620 1089
Google Scholar
[101] Yang L, Zhang Z, Song Y, Hong S, Xu R, Zhao Y, Shao Y, Zhang W, Cui B, Yang M H 2022 arXiv: 2209.00796 [cs.LG]
[102] Croitoru F A, Hondru V, Ionescu R T, Shah M 2023 IEEE Trans. Pattern Anal. Mach. Intell. 45 10850
Google Scholar
[103] Kong Z, Ping W, Huang J, Zhao K, Catanzaro B 2020 arXiv: 2009.09761 [eess.AS]
[104] Liu Y, Chen L, Liu H 2022 bioRxiv 2022.12.17.52084 Liu Y, Chen L, Liu H 2022 bioRxiv 2022.12.17.52084
[105] Watson J L, Juergens D, Bennett N R, et al. 2022 bioRxiv 2022.12.09.519842
[106] Xiong P, Wang M, Zhou X, Zhang T, Zhang J, Chen Q, Liu H 2014 Nat. Commun. 5 5330
Google Scholar
[107] Xiong P, Hu X, Huang B, Zhang J, Chen Q, Liu H 2020 Bioinformatics 36 136
Google Scholar
[108] Dauparas J, Anishchenko I, Bennett N, et al. 2022 Science 378 49
Google Scholar
[109] Zhou J, Cui G, Hu S, Zhang Z, Yang C, Liu Z, Wang L, Li C, Sun M 2020 AI open 1 57
Google Scholar
[110] Chen Y, Chen Q, Liu H 2022 J. Chem. Inf. Model. 62 971
Google Scholar
[111] Marchand A, Van Hall-Beauvais A K, Correia B E 2022 Curr. Opin. Struct. Biol. 74 102370
Google Scholar
[112] Shi C, Wang C, Lu J, Zhong B, Tang J 2022 arXiv: 2210.08761 [q-bio. BM]
[113] Dixit R, Khambhati K, Supraja K V, Singh V, Lederer F, Show P L, Awasthi M K, Sharma A, Jain R 2022 Bioresour. Technol. 128522
Google Scholar
[114] Kaptan S, Vattulainen I 2022 Adv. Phys.: X 7 2006080
Google Scholar
[115] Casadevall G, Duran C, Osuna S 2023 JACS Au 3 1554
Google Scholar
[116] Webb C, Ip S, Bathula N V, et al. 2022 Mol. Pharmaceutics 19 1047
Google Scholar
[117] Mauro V P, Chappell S A 2014 Trends Mol. Med. 20 604
Google Scholar
[118] Sarkar D, Saha S 2019 J. Biosci. 44 104
Google Scholar
-

图 1 量子力学与机器学习间的相似性. 从左到右, 从上到下的图片分别是: Chignolin蛋白质在(a)无水环境和(b)有水环境下的情况, 使用SchNet模型得到的(c)可视化电荷密度和(d)局部化学势, (e)氢原子的波函数以及(f)Müller-Brown势能. 图片引自文献[42] (版权属于美国化学会)
Fig. 1. Similarity between quantum mechanics and machine learning. Images from left to right from top to bottom: Chignolin protein (a) without and (b) with the water environment, (c) visualized total charge densities and (d) local chemical potentials obtained using the SchNet model, (e) wave functions for hydrogen atom and (f) Müller-Brown potential. Reprinted with permission from Ref. [42] (Copyright 2021 American Chemical Society).
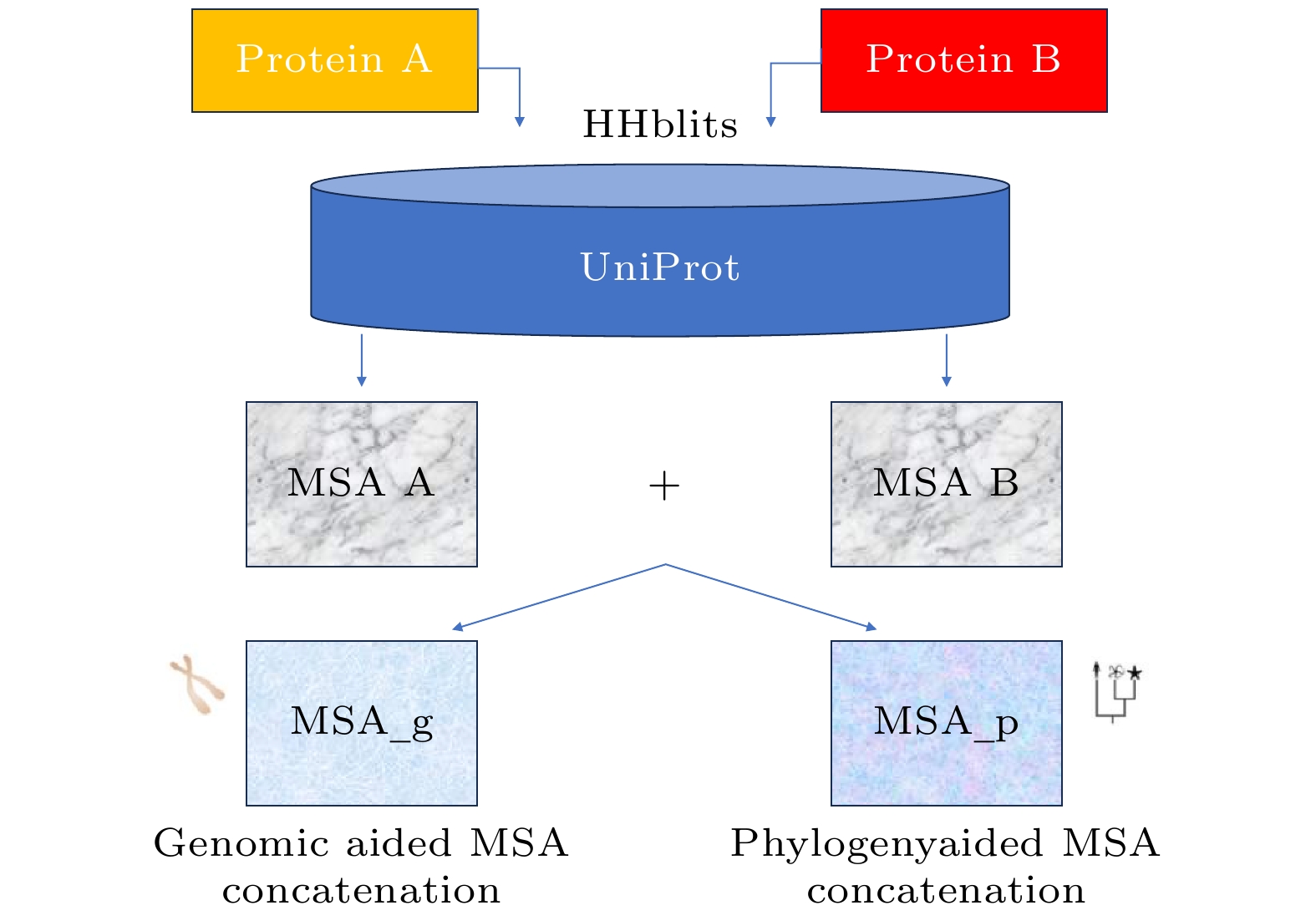
图 3 AlphaFold-Multimer的多序列比对构建方法
Fig. 3. Construction of MSA used in AlphaFold-Multimer.
-
[1] Baltoumas F A, Zafeiropoulou S, Karatzas E, et al. 2021 Biomolecules 11 1245
Google Scholar
[2] Wolf Y I, Katsnelson M I, Koonin E V 2018 Proc. Natl. Acad. Sci. USA 115 E8678
Google Scholar
[3] Fusco A, Fedele M 2007 Nat. Rev. Cancer 7 899
Google Scholar
[4] Noble D 2002 Nat. Rev. Mol. Cell Biol. 3 459
Google Scholar
[5] Markowetz F 2017 PLoS Biology 15 e2002050
Google Scholar
[6] Hollingsworth S A, Dror R O 2018 Neuron 99 1129
Google Scholar
[7] Zhang Y 2008 Curr. Opin. Struct. Biol. 18 342
Google Scholar
[8] Agostini F, Vendruscolo M, Tartaglia G G 2012 J. Mol. Biol. 421 237
Google Scholar
[9] Chen L, Fan Z, Chang J, et al. 2023 Nat. Commun. 14 4217
Google Scholar
[10] Geng H, Chen F, Ye J, Jiang F 2019 Computat. Struct. Biotechnol. J. 17 1162
Google Scholar
[11] Salo-Ahen O M, Alanko I, Bhadane R, et al. 2020 Processes 9 71
Google Scholar
[12] Norberg J, Nilsson L 2003 Q. Rev. Biophys. 36 257
Google Scholar
[13] van der Kamp M W, Shaw K E, Woods C J, Mulholland A J 2008 J. R. Soc. Interface 5 173
Google Scholar
[14] Dror R O, Dirks R M, Grossman J, Xu H, Shaw D E 2012 Annu. Rev. Biophys. 41 429
Google Scholar
[15] Lin X, Li X, Lin X 2020 Molecules 25 1375
Google Scholar
[16] Pearce R, Zhang Y 2021 Curr. Opin. Struct. Biol. 68 194
Google Scholar
[17] Jordan M I, Mitchell T M 2015 Science 349 255
Google Scholar
[18] Butler K T, Davies D W, Cartwright H, Isayev O, Walsh A 2018 Nature 559 547
Google Scholar
[19] Liakos K G, Busato P, Moshou D, Pearson S, Bochtis D 2018 Sensors 18 2674
Google Scholar
[20] Jiang T, Gradus J L, Rosellini A J 2020 Behav. Ther. 51 675
Google Scholar
[21] Hastie T, Tibshirani R, Friedman J, Hastie T, Tibshirani R, Friedman J 2009 Unsupervised Learning. In: The Elements of Statistical Learning. Springer Series in Statistics (New York: Springer) pp485–585
[22] Van Engelen J E, Hoos H H 2020 Machine Learning 109 373
Google Scholar
[23] Wiering M A, Van Otterlo M 2012 Reinforcement Learning (Heidelberg, Berlin: Springer) p729
[24] LeCun Y, Bengio Y, Hinton G 2015 Nature 521 436
Google Scholar
[25] Deng L, Yu D 2014 Deep Learning: Methods and Applications (Now Foundations and Trends) p197
[26] Jones D T 2019 Nat. Rev. Mol. Cell Biol. 20 659
Google Scholar
[27] Das P, Sercu T, Wadhawan K, et al. 2021 Nat. Biomed. Eng. 5 613
Google Scholar
[28] Kuhlman B, Bradley P 2019 Nat. Rev. Mol. Cell Biol. 20 681
Google Scholar
[29] Trevino S R, Scholtz J M, Pace C N 2008 J. Pharm. Sci. 97 4155
Google Scholar
[30] Kelley K W, Weigent D A, Kooijman R 2007 Brain Behav. Immun. 21 384
Google Scholar
[31] Babin V, Roland C, Sagui C 2008 J. Chem. Phys. 128
Google Scholar
[32] Morozov I V, Kazennov A M, Bystryi R, Norman G E, Pisarev V V, Stegailov V V 2011 Comput. Phys. Commun. 182 1974
Google Scholar
[33] Karplus M, McCammon J A 2002 Nat. Struct. Biol. 9 646
Google Scholar
[34] Wang Y, Ribeiro J M L, Tiwary P 2020 Curr. Opin. Struct. Biol. 61 139
Google Scholar
[35] Chmiela S, Tkatchenko A, Sauceda H E, Poltavsky I, Schütt K T, Müller K R 2017 Sci. Adv. 3 e1603015
Google Scholar
[36] Ponder J W, Case D A 2003 Adv. Protein Chem. 66 27
Google Scholar
[37] Monticelli L, Tieleman D P 2013 Biomolecular Simulations: Methods and Protocols 197
[38] Wang J, Wolf R M, Caldwell J W, Kollman P A, Case D A 2004 J. Comput. Chem. 25 1157
Google Scholar
[39] Hughes Z E, Wright L B, Walsh T R 2013 Langmuir 29 13217
Google Scholar
[40] Cesari A, Bottaro S, Lindorff-Larsen K, Banáš P, Šponer J, Bussi G 2019 J. Chem. Theory Comput. 15 3425
Google Scholar
[41] Unke O T, Chmiela S, Sauceda H E, Gastegger M, Poltavsky I, Schütt K T, Tkatchenko A, Müller K R 2021 Chem. Rev. 121 10142
Google Scholar
[42] Poltavsky I, Tkatchenko A 2021 J. Phys. Chem. Lett. 12 6551
Google Scholar
[43] Kästner J 2011 WIREs Comput. Mol. Sci. 1 932
Google Scholar
[44] Izrailev S, Stepaniants S, Isralewitz B, Kosztin D, Lu H, Molnar F, Wriggers W, Schulten K 1999 Computational Molecular Dynamics: Challenges, Methods, Ideas: Proceedings of the 2nd International Symposium on Algorithms for Macromolecular Modelling Berlin, May 21–24, 1997 p39
[45] Moradi M, Babin V, Roland C, Sagui C 2013 Nucleic Acids Res. 41 33
Google Scholar
[46] Simonson T, Archontis G, Karplus M 2002 Acc. Chem. Res. 35 430
Google Scholar
[47] Bitencourt-Ferreira G, de Azevedo W F 2018 Biophys. Chem. 240 63
Google Scholar
[48] Trott O, Olson A J 2010 J. Comput. Chem. 31 455
Google Scholar
[49] Besora M, Vidossich P, Lledos A, Ujaque G, Maseras F 2018 J. Phys. Chem. A 122 1392
Google Scholar
[50] Pan X, Yang J, Van R, Epifanovsky E, Ho J, Huang J, Pu J, Mei Y, Nam K, Shao Y 2021 J. Chem. Theory Comput. 17 5745
Google Scholar
[51] Senn H M, Thiel W 2009 Angew. Chem. Int. Ed. 48 1198
Google Scholar
[52] Riniker S 2017 J. Chem. Inf. Model. 57 726
Google Scholar
[53] Bennett W D, He S, Bilodeau C L, Jones D, Sun D, Kim H, Allen J E, Lightstone F C, Ingólfsson H I 2020 J. Chem. Inf. Model. 60 5375
Google Scholar
[54] Bertazzo M, Gobbo D, Decherchi S, Cavalli A 2021 J. Chem. Theory Comput. 17 5287
Google Scholar
[55] Eswar N, John B, Mirkovic N, et al. 2003 Nucleic Acids Research 31 3375
Google Scholar
[56] Asara J M, Schweitzer M H, Freimark L M, Phillips M, Cantley L C 2007 Science 316 280
Google Scholar
[57] Greener J G, Kandathil S M, Moffat L, Jones D T 2022 Nat. Rev. Mol. Cell Biol. 23 40
Google Scholar
[58] Jumper J, Evans R, Pritzel A, et al. 2021 Nature 596 583
Google Scholar
[59] Wu R, Ding F, Wang R, et al. 2022 bioRxiv 2022.07.21. 500999
[60] Baek M, DiMaio F, Anishchenko I, et al. 2021 Science 373 871
Google Scholar
[61] Medsker L R, Jain L 1999 Recurrent Neural Networks: Design and Applications (1st Ed.) (CRC Press) p2
[62] Kim P 2017 Convolutional Neural Network. In: MATLAB Deep Learning (Berkeley, CA: Apress) p121
[63] Wardah W, Khan M G, Sharma A, Rashid M A 2019 Comput. Biol. Chem. 81 1
Google Scholar
[64] Mirabello C, Pollastri G 2013 Bioinformatics 29 2056
Google Scholar
[65] Heffernan R, Yang Y, Paliwal K, Zhou Y 2017 Bioinformatics 33 2842
Google Scholar
[66] Wang S, Peng J, Ma J, Xu J 2016 Sci. Rep. 6 1
Google Scholar
[67] Li Z, Yu Y 2016 arXiv: 1604.07176 [q-bio.BM]
[68] Wang Y, Mao H, Yi Z 2017 Knowledge-Based Systems 118 115
Google Scholar
[69] Nishikawa K, Ooi T, Isogai Y, Saitô N 1972 J. Phys. Soc. JPN 32 1331
Google Scholar
[70] Edgar R C, Batzoglou S 2006 Curr. Opin. Struct. Biol. 16 368
Google Scholar
[71] Vaswani A, Shazeer N, Parmar N, Uszkoreit J, Jones L, Gomez A N, Kaiser Ł, Polosukhin I 2017 Advances in Neural Information Processing Systems 30 Long Beach, USA, December 4–9, 2017 p30
[72] Janin J, Bahadur R P, Chakrabarti P 2008 Q. Rev. Biophys. 41 133
Google Scholar
[73] Zafferani M, Hargrove A E 2021 Cell Chem. Biol. 28 594
Google Scholar
[74] Hunter C A 2004 Angew. Chem. Int. Ed. 43 5310
Google Scholar
[75] Chen R, Li L, Weng Z 2003 Proteins Struct. Funct. Bioinf. 52 80
Google Scholar
[76] Jingcheng Y, Zhaoming C, Zhaoqun L, Mingliang Z, Wenjun L, He H, Qiwei Y 2022 Code of Open Complex https:// github.com/baaihealth/OpenComplex.
[77] Evans R, O’ Neill M, Pritzel A, et al. 2021 bioRxiv 2021.10.04.463034
[78] Moriwaki Y 2021 Twitter https://twitter.com/Ag_smith/ status.
[79] Ko J, Lee J 2021 bioRxiv 2021.07.27.453972 Ko J, Lee J 2021 bioRxiv 2021.07.27.453972
[80] Tsaban T, Varga J K, Avraham O, Ben-Aharon Z, Khramushin A, Schueler-Furman O 2022 Nat. Commun. 13 176
Google Scholar
[81] Bryant P, Pozzati G, Elofsson A 2022 Nat. Commun. 13 1265
Google Scholar
[82] Zhou T M, Wang S, Xu J 2017 bioRxiv 240754
[83] Cang Z, Wei G W 2017 PLoS Comput. Biol. 13 e1005690
Google Scholar
[84] Yagi K, Re S, Mori T, Sugita Y 2022 Curr. Opin. Struct. Biol. 72 88
Google Scholar
[85] Vendruscolo M, Knowles T P, Dobson C M 2011 CSH Perspect. Biol. 3 a010454
Google Scholar
[86] Khurana S, Rawi R, Kunji K, Chuang G Y, Bensmail H, Mall R 2018 Bioinformatics 34 2605
Google Scholar
[87] Wu X, Yu L 2021 Bioinformatics 37 4314
Google Scholar
[88] Schellekens H 2003 Nephrology Dialysis Transplantation 18 1257
Google Scholar
[89] Ternette N, Tippler B, Überla K, Grunwald T 2007 Vaccine 25 7271
Google Scholar
[90] Jefferis R 2016 J. Immunol. Res. 2016
Google Scholar
[91] Schellekens H 2005 Nephrology Dialysis Transplantation 20 vi3
Google Scholar
[92] Smith C C, Chai S, Washington A R, et al. 2019 Cancer Immunol. Res. 7 1591
Google Scholar
[93] Gonzalez-Dias P, Lee E K, Sorgi S, de Lima D S, Urbanski A H, Silveira E L, Nakaya H I 2020 Hum. Vacc. Immunother. 16 269
Google Scholar
[94] Timr S, Madern D, Sterpone F 2020 Prog. Mol. Biol. Transl. Sci. 170 239
Google Scholar
[95] Pudžiuvelytė I, Olechnovič K, Godliauskaite E, Sermokas K, Urbaitis T, Gasiunas G, Kazlauskas D 2023 bioRxiv 2023.03.27.534365 Pudžiuvelytė I, Olechnovič K, Godliauskaite E, Sermokas K, Urbaitis T, Gasiunas G, Kazlauskas D 2023 bioRxiv 2023.03.27.534365
[96] Rives A, Meier J, Sercu T, et al. 2021 Proc. Natl. Acad. Sci. U.S.A. 118 e2016239118
Google Scholar
[97] Elnaggar A, Heinzinger M, Dallago C, et al. 2022 IEEE Trans. Pattern Anal. Mach. Intell. 44 7112
Google Scholar
[98] Huang P S, Boyken S E, Baker D 2016 Nature 537 320
Google Scholar
[99] Huang B, Xu Y, Hu X, Liu Y, Liao S, Zhang J, Huang C, Hong J, Chen Q, Liu H 2022 Nature 602 523
Google Scholar
[100] Watson J L, Juergens D, Bennett N R, et al. 2023 Nature 620 1089
Google Scholar
[101] Yang L, Zhang Z, Song Y, Hong S, Xu R, Zhao Y, Shao Y, Zhang W, Cui B, Yang M H 2022 arXiv: 2209.00796 [cs.LG]
[102] Croitoru F A, Hondru V, Ionescu R T, Shah M 2023 IEEE Trans. Pattern Anal. Mach. Intell. 45 10850
Google Scholar
[103] Kong Z, Ping W, Huang J, Zhao K, Catanzaro B 2020 arXiv: 2009.09761 [eess.AS]
[104] Liu Y, Chen L, Liu H 2022 bioRxiv 2022.12.17.52084 Liu Y, Chen L, Liu H 2022 bioRxiv 2022.12.17.52084
[105] Watson J L, Juergens D, Bennett N R, et al. 2022 bioRxiv 2022.12.09.519842
[106] Xiong P, Wang M, Zhou X, Zhang T, Zhang J, Chen Q, Liu H 2014 Nat. Commun. 5 5330
Google Scholar
[107] Xiong P, Hu X, Huang B, Zhang J, Chen Q, Liu H 2020 Bioinformatics 36 136
Google Scholar
[108] Dauparas J, Anishchenko I, Bennett N, et al. 2022 Science 378 49
Google Scholar
[109] Zhou J, Cui G, Hu S, Zhang Z, Yang C, Liu Z, Wang L, Li C, Sun M 2020 AI open 1 57
Google Scholar
[110] Chen Y, Chen Q, Liu H 2022 J. Chem. Inf. Model. 62 971
Google Scholar
[111] Marchand A, Van Hall-Beauvais A K, Correia B E 2022 Curr. Opin. Struct. Biol. 74 102370
Google Scholar
[112] Shi C, Wang C, Lu J, Zhong B, Tang J 2022 arXiv: 2210.08761 [q-bio. BM]
[113] Dixit R, Khambhati K, Supraja K V, Singh V, Lederer F, Show P L, Awasthi M K, Sharma A, Jain R 2022 Bioresour. Technol. 128522
Google Scholar
[114] Kaptan S, Vattulainen I 2022 Adv. Phys.: X 7 2006080
Google Scholar
[115] Casadevall G, Duran C, Osuna S 2023 JACS Au 3 1554
Google Scholar
[116] Webb C, Ip S, Bathula N V, et al. 2022 Mol. Pharmaceutics 19 1047
Google Scholar
[117] Mauro V P, Chappell S A 2014 Trends Mol. Med. 20 604
Google Scholar
[118] Sarkar D, Saha S 2019 J. Biosci. 44 104
Google Scholar

 下载:
下载:

计量
- 文章访问数: 8261
- PDF下载量: 168
- 被引次数: 0
