










-
Using solar photoelectrochemical decomposition of water to produce hydrogen and oxygen is one of the most feasible approaches to obtaining renewable energy. Compared with hydrogen-evolution reaction (HER), the oxygen-evolution reaction (OER) is very complex, there are four sluggish proton-coupled electron transfer processes. It is critical to improve OER performance. The BiVO4 (010) facet possesses low surface energy, strong visible absorption, and good activity for OER, and is considered as one of the most suitable PEC catalysts. However, its poor electron conductivity, low charge carrier mobility, and high charge recombination rates significantly limit its practical applications. To achieve highly active OER photocatalysts, we modify BiVO4 (010) facet by substitutial doping with Al atom and surface adsorption with Al atom. According to density functional theory calculations, we compare OER performances of these two modified BiVO4 (010) facets. The results show that both approaches can effectively regulate the electronic structure of BiVO4 and then tune OER activity resulting from the change of the structure. Though Al substitutional doping reduces the band gap of the (010) facet and enhances the visible light absorption, the improvement of OER performance is not significant because the doping site is inside and has little influence on the surface active site. Importantly, the surface adsorption of Al atom is considered as an efficient means to improve the OER activity on BiVO4 (010) facet due to the combined action between surface adsorbed Al and active site Bi atoms. Al adsorbed (010) facet exhibits excellent OER catalytic activity: 1) the induction of localized states and the reduction of band gap are conducive to the electronic transition, optical absorption, thus increasing the electrical conductivity; 2) there is lower hole effective mass, and thus effectively enhancing the ability to transfer from anode surface to electrolyte surface, thereby increasing the difference between the effective mass ratio of electron−hole pairs and 1 and effectively reducing the electron-hole recombination; 3) the nteraction between the active sites and oxygen-containing intermediates is reinforced in the OER process, therefore the potential determining step of OER decreases effectively. This work provides an important reference for designing efficient and stable two-dimensional semiconductor-based photocatalysts for OER. We believe that it will arouse great interest of the BiVO4 community and motivate numerous experimental researches.
-
Keywords:
- water-splitting /
- substitutional doping /
- surface adsorption /
- oxygen evolution reaction /
- BiVO4
[1] Pavone M, Caspary Toroker M 2020 ACS Energy Lett. 5 2042
 Google Scholar
Google Scholar
[2] Tahir M B, Nawaz T, Nabi G, Sagir M, Rafique M, Ahmed A, Muhammad S 2020 Int. J. Hydrogen Energy 45 22833
Google Scholar
[3] Mushtaq M A, Arif M, Fang X, Yasin G, Ye W, Basharat M, Zhou B, Yang S, Ji S, Yan D 2021 J. Mater. Chem. A 9 2742
Google Scholar
[4] Han H, Kment S, Karlicky F, Wang L, Naldoni A, Schmuki P, Zboril R 2018 Small 14 1
[5] Huang H, Shang M, Zou Y, Song W, Zhang Y 2019 Nanoscale 11 21188
Google Scholar
[6] Tian C M, Li W W, Lin Y M, et al. 2020 J. Phys. Chem. C 124 12548
Google Scholar
[7] Kudo A, Miseki Y 2009 Chem. Soc. Rev. 38 253
Google Scholar
[8] Thalluri S M, Bai L, Lv C, Huang Z, Hu X, Liu L 2020 Adv. Sci. 7 1902102
Google Scholar
[9] Pan J, Ma X, Zhang W, Hu J 2021 RSC Adv. 12 540
[10] Thalluri S M, Suarez C M, Hussain M, Hernandez S, Virga A, Saracco G, Russo N 2013 Ind. Eng. Chem. Res. 52 17414
Google Scholar
[11] Wang Q, Lin Y, Li P, Ma M, Maheskumar V, Jiang Z, Zhang R 2021 Int. J. Hydrogen Energy 46 247
Google Scholar
[12] Li P, Chen X, He H, Zhou X, Zhou Y, Zou Z 2018 Adv. Mater. 30 4
[13] Irani R, Ahmet I Y, Jang J W, et al. 2020 Sol. Rrl. 4 1900290
Google Scholar
[14] Massaro A, Pecoraro A, Hernández S, Talarico G, Muñoz-García A B, Pavone M 2022 Mol. Catal. 517 112036
Google Scholar
[15] Qi Y, Zhang J, Kong Y, et al. 2022 Nat. Commun. 13 1
[16] Wen L, Ding K, Huang S, Zhang Y, Li Y, Chen W 2017 New J. Chem. 41 1094
Google Scholar
[17] Ullah H, Tahir A A, Mallick T K 2018 Appl. Catal. B Environ. 224 895
Google Scholar
[18] Maheskumar V, Lin Y M, Jiang Z, Vidhya B, Ghosal A 2022 J. Photochem. Photobiol. A Chem. 426 113757
Google Scholar
[19] Zhao X, Hu J, Yao X, Chen S, Chen Z 2018 ACS Appl. Energy Mater. 1 3410
Google Scholar
[20] Ma L, Liu Z, Chen T, Liu Y, Fang G 2020 Electrochim. Acta 355 136777
Google Scholar
[21] Kresse G, Furthmüller J 1996 Comput. Mater. Sci. 6 15
Google Scholar
[22] Kresse G, Hafner J 1994 Phys. Rev. B 49 14251
Google Scholar
[23] Perdew J P, Burke K, Ernzerhof M 1996 Phys. Rev. Lett. 77 3865
Google Scholar
[24] Blöchl P E 1994 Phys. Rev. B 50 17953
Google Scholar
[25] Hu J, Zhao X, Chen W, Su H, Chen Z 2017 J. Phys. Chem. C 121 18702
Google Scholar
[26] Tokunaga S, Kato H, Kudo A 2001 Chem. Mater. 13 4624
Google Scholar
[27] Kahneman D, Tversky A 1979 Asp. Gen. La Planif. Tribut. En Venez. 2009 31
[28] Zhang X, Huang Y, Ma F, Zhang Z, Wei X 2018 J. Phys. Chem. Solids 121 85
Google Scholar
[29] Anke B, Rohloff M, Willinger M G, Hetaba W, Fischer A, Lerch M 2017 Solid State Sci. 63 1
Google Scholar
[30] Zhao R, Zhang L, Fan G, Chen Y, Huang G, Zhang H, Zhu J, Guan X 2021 Cem. Concr. Res. 144 106420
Google Scholar
[31] Würfel P 2003 Sol. Energy Mater. Sol. Cells 79 153
Google Scholar
[32] Ma H, Chen X Q, Li R, Wang S, Dong J, Ke W 2017 Acta Mater. 130 137
Google Scholar
[33] Shi J, Zhang W, Gu Q 2022 Solid State Commun. 351 114794
Google Scholar
[34] Bi Y, Yang Y, Shi X L, Feng L, Hou X, Ye X, Zhang L, Suo G, Lu S, Chen Z G 2021 J. Mater. Sci. Technol. 83 102
Google Scholar
-
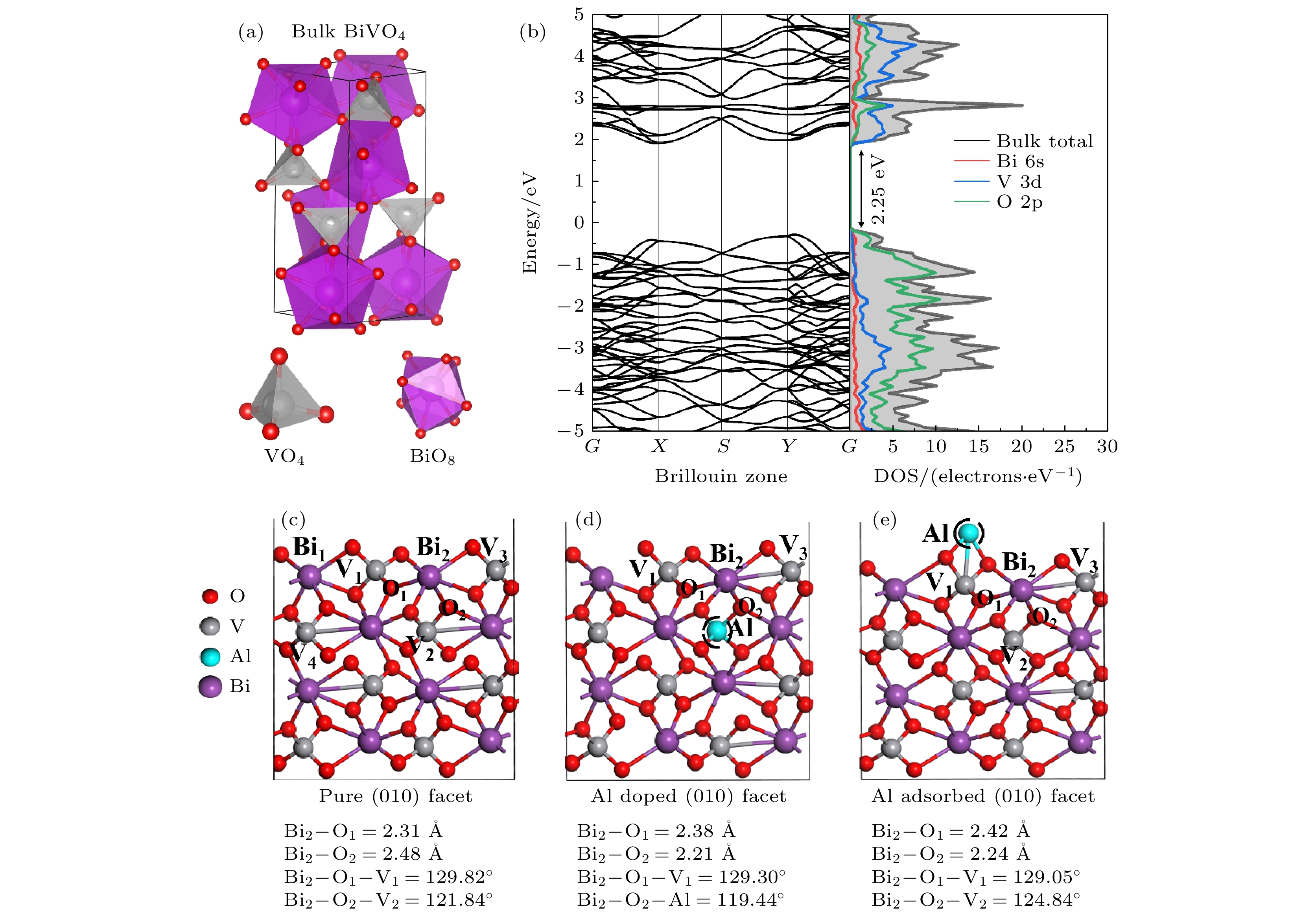
图 1 (a) ms-BiVO4块体结构侧视图; (b) 块体钒酸铋的能带结构与态密度; (c) BiVO4 (010)面结构的侧视图; (d) Al替代V位点的侧视图; (e) Al原子吸附在BiVO4 (010)晶面的侧视图; BiVO4 (010)晶面共48个原子, 包括8个Bi (紫色)、8个V (灰色)和32个O (红色)原子
Fig. 1. (a) The side view of bulk ms-BiVO4; (b) band structure and PDOS of bulk BiVO4; the side views of (c) pristine, (d) Al doped and (e) Al adsorbed BiVO4 (010) facets. There are 48 atoms in BiVO4 (010) facet including 8 Bi (purple), 8 V (gray), and 32 O (red) atoms.
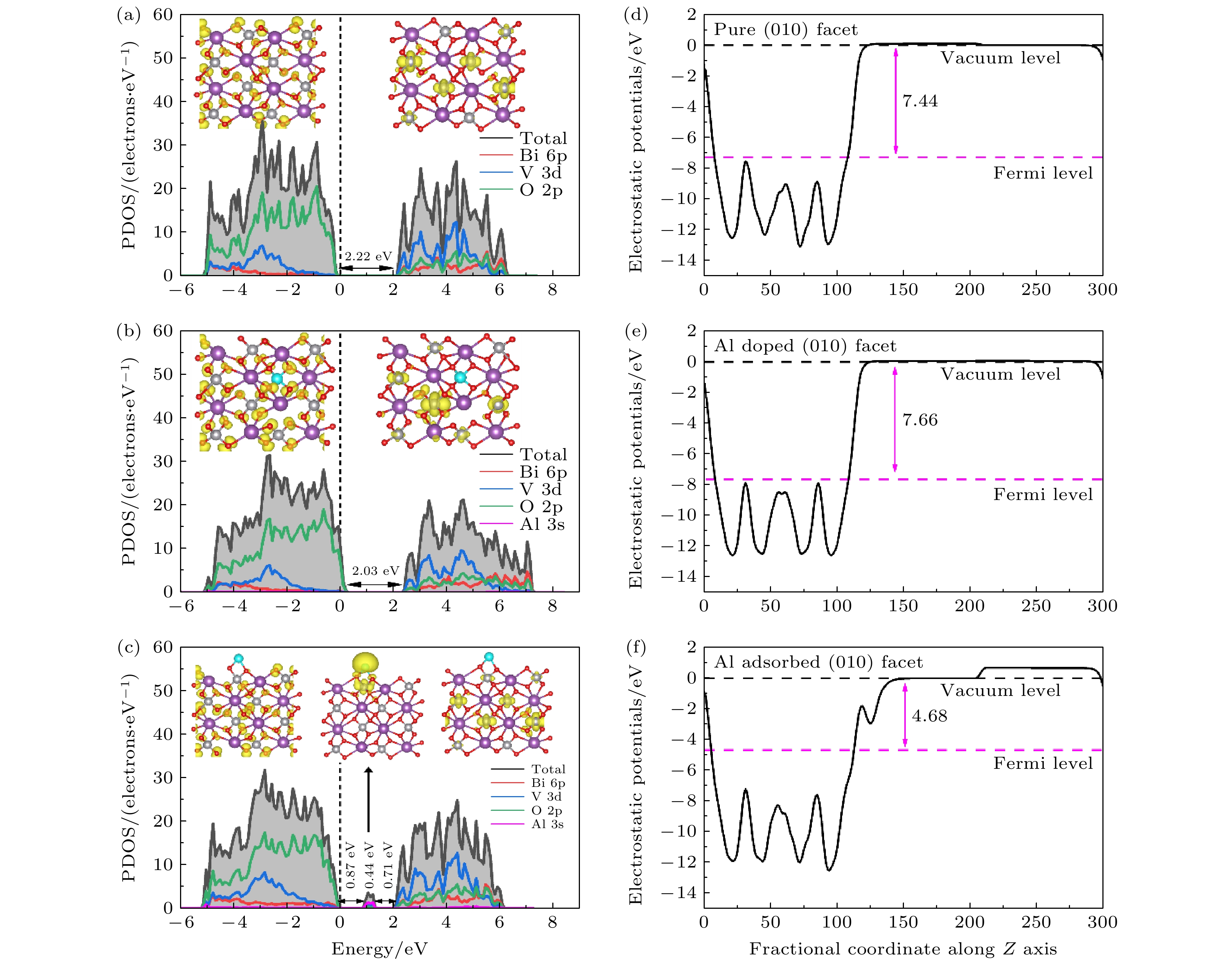
图 2 (a) 原始的、(b) Al替位掺杂和(c) 表面吸附的BiVO4 (010)晶面总态密度和分波态密度; 插图是导带底和价带顶处电荷密度图, 费米能级设置为零; (d) 原始的、(e) Al替位掺杂和(f) 表面吸附BiVO4 (010)晶表面沿 z 轴的平均静电势
Fig. 2. The total and partial density of states of (a) pure, (b) Al substitutional doped and (c) surface adsorbed BiVO4 (010) surfaces; the inset is the charge density of VBM and CBM , the Fermi level is set to zero. Average electrostatic potentials along the z axis of (d) pure, (e) Al substitutional doped and (f) surface adsorbed BiVO4 (010) surfaces.
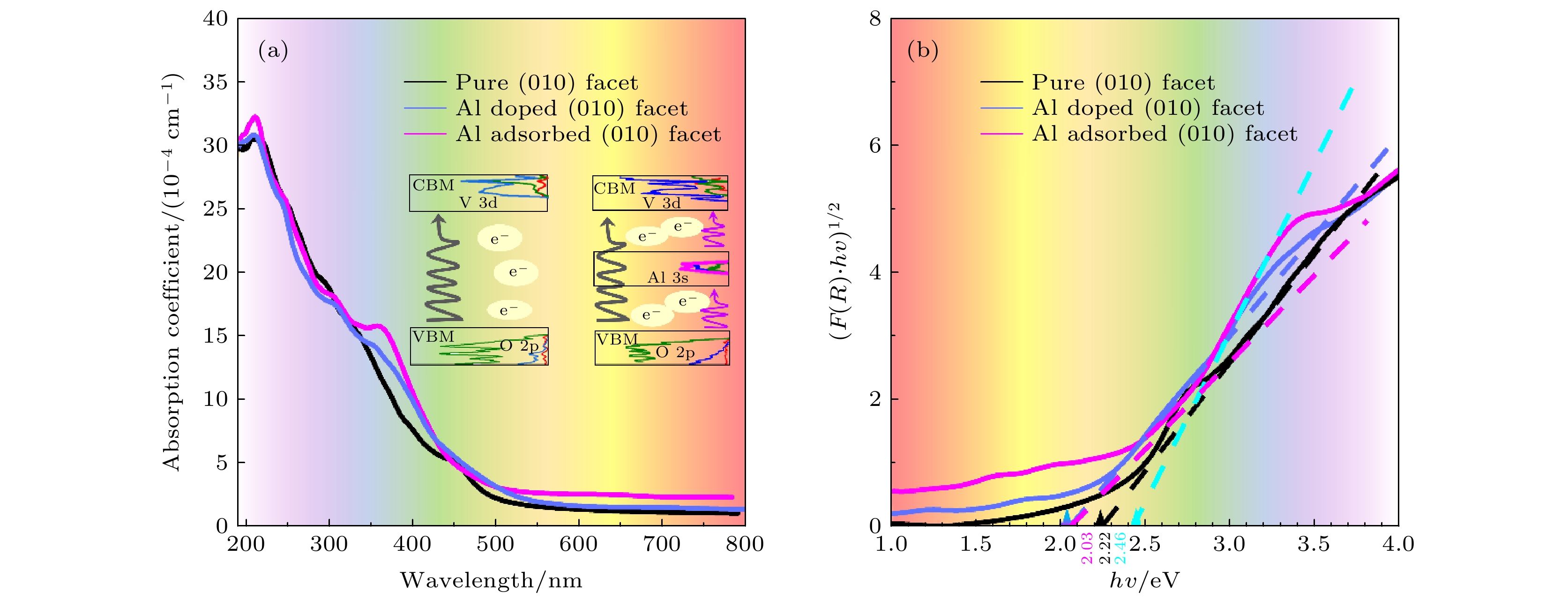
图 3 原始的、Al替位掺杂和表面吸附BiVO4的(010)晶面的(a)光吸收谱图; (b) (F(R)·hν)1/2与光子能量关系图像
Fig. 3. (a) The calculated absorption coefficient and (b) the (F(R)·hν)1/2 with the change of photon energy in pure, Al substitutional doped and surface adsorbed BiVO4 (010) surfaces.

图 4 OER四电子步过程中含氧中间体H2Oads, HOads, Oads和HOOads吸附在原始的、Al替位掺杂和表面吸附的BiVO4 (010)表面以Bi或Al为活性位点的吸附结构和吸附能. “–”和“@”符号分别表示(010)面上的键和吸附状态、吸附能与键长的统一单位为eV和Å
Fig. 4. The adsorbed structure and adsorbed energies of the oxygenated intermediates of H2Oads, HOads, Oads and HOOads adsorbed on pure, Al substitutional doped and surface adsorbed BiVO4 (010) surfaces during the four steps of OER, where Bi and Al respectively act as active site. The “–”, and “@” signs stand for bond, and adsorption state on the surface, the unity units of adsorption energy and bond length are eV and Å, respectively.
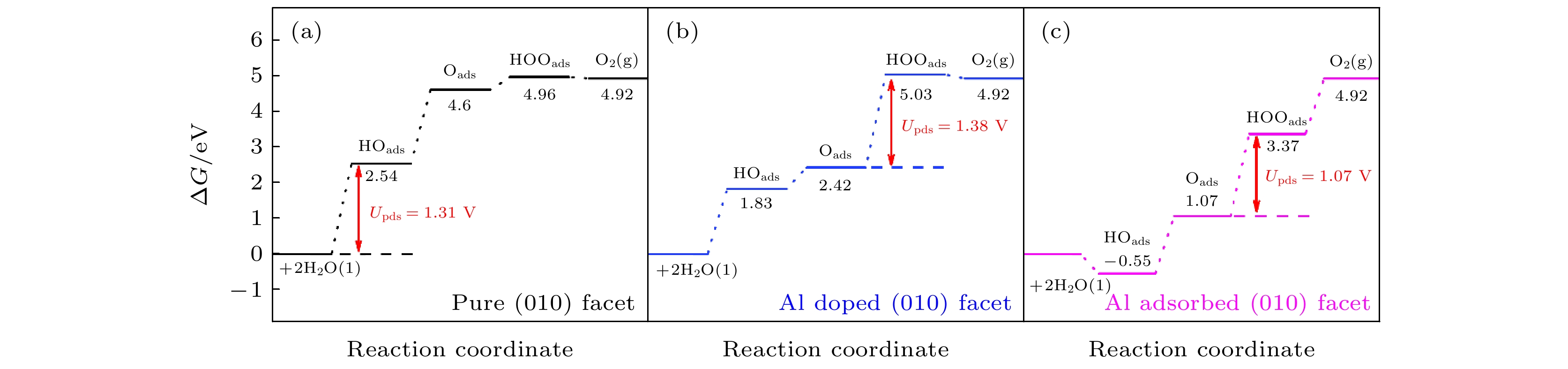
图 5 OER四个电子步在U = 0, pH = 0, T = 298 K下自由能台阶图 (a)原始BiVO4 (010)晶面; (b) Al替位掺杂(010)晶面; (c) 表面吸附(010)晶面, Upds表示决速步的过电势
Fig. 5. Free energy profiles of OER on (a) pure BiVO4 (010) facet; (b) Al doped (010) facet and (c) Al adsorbed (010) facet at U = 0, pH = 0, T = 298 K. Upds represents the potential of the rate determining step.
表 1 原始的及Al原子替位掺杂和表面吸附的钒酸铋(010)晶面的形成能、禁带宽度、功函数、电子有效质量、空穴有效质量、电子-空穴有效质量比、决速步过电势
Table 1. The formation energy, band gap, the work function, effective mass of electron, effective mass of hole and relative ratio of the effective masses, the potential of the rate determining step of pure, Al substitutional doped and surface adsorbed BiVO4 (010) surfaces.
System Eform/eV Eg /eV W⊥/eV $m_{\rm{e}}^* $/me $m_{\rm{h}}^* $/me D Upds/V Pure (010) facet 2.22 7.44 1.55 1.49 1.04 1.31 Al doped (010) facet –6.32 2.03 7.66 1.53 7.97 0.19 1.38 Al adsorbed (010) facet –2.92 2.02 4.68 1.90 0.41 4.63 1.07  下载: 导出CSV
下载: 导出CSV
-
[1] Pavone M, Caspary Toroker M 2020 ACS Energy Lett. 5 2042
Google Scholar
[2] Tahir M B, Nawaz T, Nabi G, Sagir M, Rafique M, Ahmed A, Muhammad S 2020 Int. J. Hydrogen Energy 45 22833
Google Scholar
[3] Mushtaq M A, Arif M, Fang X, Yasin G, Ye W, Basharat M, Zhou B, Yang S, Ji S, Yan D 2021 J. Mater. Chem. A 9 2742
Google Scholar
[4] Han H, Kment S, Karlicky F, Wang L, Naldoni A, Schmuki P, Zboril R 2018 Small 14 1
[5] Huang H, Shang M, Zou Y, Song W, Zhang Y 2019 Nanoscale 11 21188
Google Scholar
[6] Tian C M, Li W W, Lin Y M, et al. 2020 J. Phys. Chem. C 124 12548
Google Scholar
[7] Kudo A, Miseki Y 2009 Chem. Soc. Rev. 38 253
Google Scholar
[8] Thalluri S M, Bai L, Lv C, Huang Z, Hu X, Liu L 2020 Adv. Sci. 7 1902102
Google Scholar
[9] Pan J, Ma X, Zhang W, Hu J 2021 RSC Adv. 12 540
[10] Thalluri S M, Suarez C M, Hussain M, Hernandez S, Virga A, Saracco G, Russo N 2013 Ind. Eng. Chem. Res. 52 17414
Google Scholar
[11] Wang Q, Lin Y, Li P, Ma M, Maheskumar V, Jiang Z, Zhang R 2021 Int. J. Hydrogen Energy 46 247
Google Scholar
[12] Li P, Chen X, He H, Zhou X, Zhou Y, Zou Z 2018 Adv. Mater. 30 4
[13] Irani R, Ahmet I Y, Jang J W, et al. 2020 Sol. Rrl. 4 1900290
Google Scholar
[14] Massaro A, Pecoraro A, Hernández S, Talarico G, Muñoz-García A B, Pavone M 2022 Mol. Catal. 517 112036
Google Scholar
[15] Qi Y, Zhang J, Kong Y, et al. 2022 Nat. Commun. 13 1
[16] Wen L, Ding K, Huang S, Zhang Y, Li Y, Chen W 2017 New J. Chem. 41 1094
Google Scholar
[17] Ullah H, Tahir A A, Mallick T K 2018 Appl. Catal. B Environ. 224 895
Google Scholar
[18] Maheskumar V, Lin Y M, Jiang Z, Vidhya B, Ghosal A 2022 J. Photochem. Photobiol. A Chem. 426 113757
Google Scholar
[19] Zhao X, Hu J, Yao X, Chen S, Chen Z 2018 ACS Appl. Energy Mater. 1 3410
Google Scholar
[20] Ma L, Liu Z, Chen T, Liu Y, Fang G 2020 Electrochim. Acta 355 136777
Google Scholar
[21] Kresse G, Furthmüller J 1996 Comput. Mater. Sci. 6 15
Google Scholar
[22] Kresse G, Hafner J 1994 Phys. Rev. B 49 14251
Google Scholar
[23] Perdew J P, Burke K, Ernzerhof M 1996 Phys. Rev. Lett. 77 3865
Google Scholar
[24] Blöchl P E 1994 Phys. Rev. B 50 17953
Google Scholar
[25] Hu J, Zhao X, Chen W, Su H, Chen Z 2017 J. Phys. Chem. C 121 18702
Google Scholar
[26] Tokunaga S, Kato H, Kudo A 2001 Chem. Mater. 13 4624
Google Scholar
[27] Kahneman D, Tversky A 1979 Asp. Gen. La Planif. Tribut. En Venez. 2009 31
[28] Zhang X, Huang Y, Ma F, Zhang Z, Wei X 2018 J. Phys. Chem. Solids 121 85
Google Scholar
[29] Anke B, Rohloff M, Willinger M G, Hetaba W, Fischer A, Lerch M 2017 Solid State Sci. 63 1
Google Scholar
[30] Zhao R, Zhang L, Fan G, Chen Y, Huang G, Zhang H, Zhu J, Guan X 2021 Cem. Concr. Res. 144 106420
Google Scholar
[31] Würfel P 2003 Sol. Energy Mater. Sol. Cells 79 153
Google Scholar
[32] Ma H, Chen X Q, Li R, Wang S, Dong J, Ke W 2017 Acta Mater. 130 137
Google Scholar
[33] Shi J, Zhang W, Gu Q 2022 Solid State Commun. 351 114794
Google Scholar
[34] Bi Y, Yang Y, Shi X L, Feng L, Hou X, Ye X, Zhang L, Suo G, Lu S, Chen Z G 2021 J. Mater. Sci. Technol. 83 102
Google Scholar

 下载:
下载:
计量
- 文章访问数: 8606
- PDF下载量: 132
- 被引次数: 0
