










-
掺氮石墨烯具有良好的应用前景, 但对其摩擦学特性的研究仍较为缺乏. 本文采用分子动力学方法研究了氮掺杂对石墨烯摩擦学特性的影响. 结果表明在公度、非公度的界面结构下, 氮掺杂对石墨烯摩擦特性的影响呈现相反的趋势. 界面结构为公度状态时, 氮原子的引入导致了局部非公度状态, 因而界面势垒降低、摩擦减小. 界面公度性的改变、层间氮原子和碳原子的范德瓦耳斯力作用对界面摩擦的影响相反, 在二者的共同作用下, 随氮掺杂比例的升高, 掺氮石墨烯体系的界面摩擦力呈现先增大再减小的趋势. 界面结构为非公度状态时, 氮原子的引入对界面摩擦的影响主要体现在原子类型的变化, 界面摩擦随氮掺杂比例的增大而增大. 存在空位缺陷的石墨烯体系的摩擦最大, 掺杂氮原子对于降低缺陷石墨烯体系的摩擦具有积极意义.Graphene has attracted a lot of attention due to its excellent electrical properties, however, the gapless nature of graphene limits its further applications. Doping is an effective way to open the bandgap, in which nitrogen-doped (N-doped) graphene has potential applications, but the study of its tribological properties is still lacking. In this work, the effects of nitrogen doping on the tribological properties of graphene under different interfacial structures are investigated by molecular dynamics simulation. The simulation models include a hexagonal graphene sheet, graphene or N-doped graphene substrate. The results show that the nitrogen doping has different effects on friction when interface structure is in a commensurate state and an incommensurate state. In a commensurate state, N-doping reduces the friction between interfaces in all cases, but the friction first goes up and then decreases with the increase of doping ratio of nitrogen. The local maximum value of friction occurs at a doping ratio of 7.5%. This phenomenon results from the interface structure and the change of van de Waals force between interfaces. The introduction of nitrogen atoms causes the lattice of graphene to distort, which results in the formation of local incommensurate state, thus reducing the interfacial potential barrier and friction. However, the van der Waals force between nitrogen atom and carbon atom between layers is stronger than that between carbon atoms and carbon atoms, which causes the friction to increase. When the doping ratio is low or high, lattice distortion plays more important role. The friction of N-doped graphene shows much smaller increase with load than that of ideal graphene, which indicates that the N-doped graphene possesses a better performance under high load. When the interface structure is in an incommensurate state, the introduction of nitrogen atoms shows slight influence on lattice mismatch, therefore, the change of atomic type plays a dominant role in determining the friction between interfaces, which goes up with the increase of N-doping ratio. When the substrate is graphene with vacancy defects, the value of friction between interfaces is larger than the ideal graphene substrate or N-doped graphene substrate, which indicates that the doping of nitrogen atoms has positive effect on reducing the friction of graphene with defects.
-
Keywords:
- graphene /
- N-doped /
- friction /
- molecular dynamics simulation
[1] Novoselov K S, Geim A K, Morozov S V, Jiang D, Zhang Y, Dubonos S V, Grigorieva I V, Firsov A A 2004 Science 306 666
 Google Scholar
Google Scholar
[2] Geim A K, Novoselov K S 2007 Nat. Mater. 6 183
Google Scholar
[3] Geim A K 2009 Science 324 1530
Google Scholar
[4] Martins T B, Miwa R H, da Silva A J R, Fazzio A 2007 Phys. Rev. Lett. 98 196803
Google Scholar
[5] Dutta S, Manna A K, Pati S K 2009 Phys. Rev. Lett. 102 096601
Google Scholar
[6] Biel B, Blase X, Triozon F, Roche S 2009 Phys. Rev. Lett. 102 096803
Google Scholar
[7] Kamal Kandezi M, Shadman Lakmehsari M, Matta C F 2020 J. Mol. Liq. 302 112574
Google Scholar
[8] 林婷婷, 吕秋丰 2015 功能材料 46 5007
Google Scholar
Lin T T, Lv Q F 2015 J. Func. Mater 46 5007
Google Scholar
[9] Wang G, Wang B, Wang X, Park J, Dou S, Ahn H, Kim K 2009 J. Mate. Chem. 19 8378
Google Scholar
[10] Kwon O S, Park S J, Hong J-Y, Han A R, Lee J S, Lee J S, Oh J H, Jang J 2012 ACS. Nano. 6 1486
Google Scholar
[11] Dienwiebel M, Verhoeven G S, Pradeep N, et al. 2004 Phys. Rev. Lett. 92 126101
Google Scholar
[12] Zheng X, Gao L, Yao Q, et al. 2016 Nat. Commun. 7 13204
Google Scholar
[13] Leven I, Krepel D, Shemesh O, Hod O 2013 J. Phys. Chem. Lett. 4 115
Google Scholar
[14] Liu S W, Wang H P, Xu Q, et al. 2017 Nat. Commun. 8 14029
Google Scholar
[15] 陈勇, 李瑞 2019 物理学报 68 186801
Google Scholar
Chen Y, Li R 2019 Acta Phys. Sin. 68 186801
Google Scholar
[16] Wang K, Ouyang W, Cao W, Ma M, Zheng Q 2019 Nanoscale 11 2186
Google Scholar
[17] Liu Y, Grey F, Zheng Q 2014 Sci. Rep. 4 4875
Google Scholar
[18] Biswas S K 2008 Tribol Lett 30 159
Google Scholar
[19] Lee C, Li Q, Kalb W, Liu X Z, Berger H, Carpick R W, Hone J 2010 Science 328 76
Google Scholar
[20] Li S, Li Q, Carpick R W, Gumbsch P, Liu X Z, Ding X, Sun J, Li J 2016 Nature 539 541
Google Scholar
[21] Ye Z, Martini A 2014 Langmuir 30 14707
Google Scholar
[22] Guo Y, Guo W, Chen C 2007 Phys. Rev. B 76 155429
Google Scholar
[23] Dong Y, Wu X, Martini A 2013 Nanotechnology 24 375701
Google Scholar
[24] Gao L, Chen X, Yan Y, Liu Y, Song A, Ma T, Hu Y, Su Y, Qiao L 2018 Comput. Mater. Sci. 147 28
Google Scholar
[25] Li R, Song C 2018 Crystals 8 167
Google Scholar
[26] Shen Y, Wang Y, Zhou Y, Shi A, Hu J, Zhang Y 2016 J. Phys. D 49 415303
Google Scholar
[27] Stuart S J, Tutein A B, Harrison J A 2000 J. Chem. Phys. 112 6472
Google Scholar
[28] Kınacı A, Haskins J B, Sevik C, Çağın T 2012 Phys. Rev. B 86 115410
Google Scholar
[29] Lennard-Jones J E 1931 Proceed. Phys. Soc. 43 461
Google Scholar
[30] Leven I, Maaravi T, Azuri I, Kronik L, Hod O 2016 J. Chem. Theory Comput. 12 2896
Google Scholar
[31] Zhang W, Lu W C, Zhang H X, Ho K M, Wang C Z 2016 Carbon 110 330
Google Scholar
-
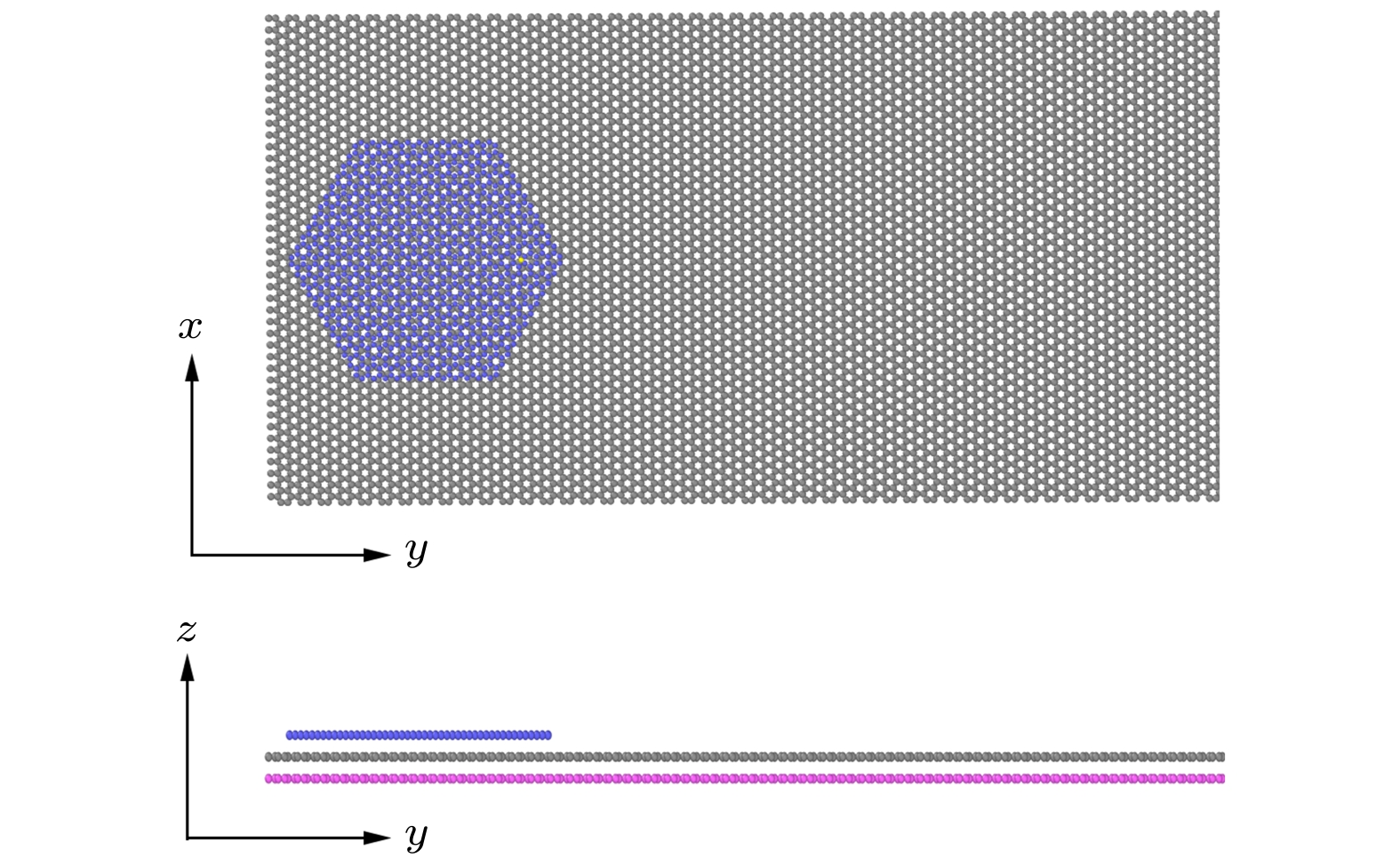
图 1 石墨烯片在石墨烯基底上的模型
Fig. 1. The illustration of hexagonal graphene sheet on graphene substrate.
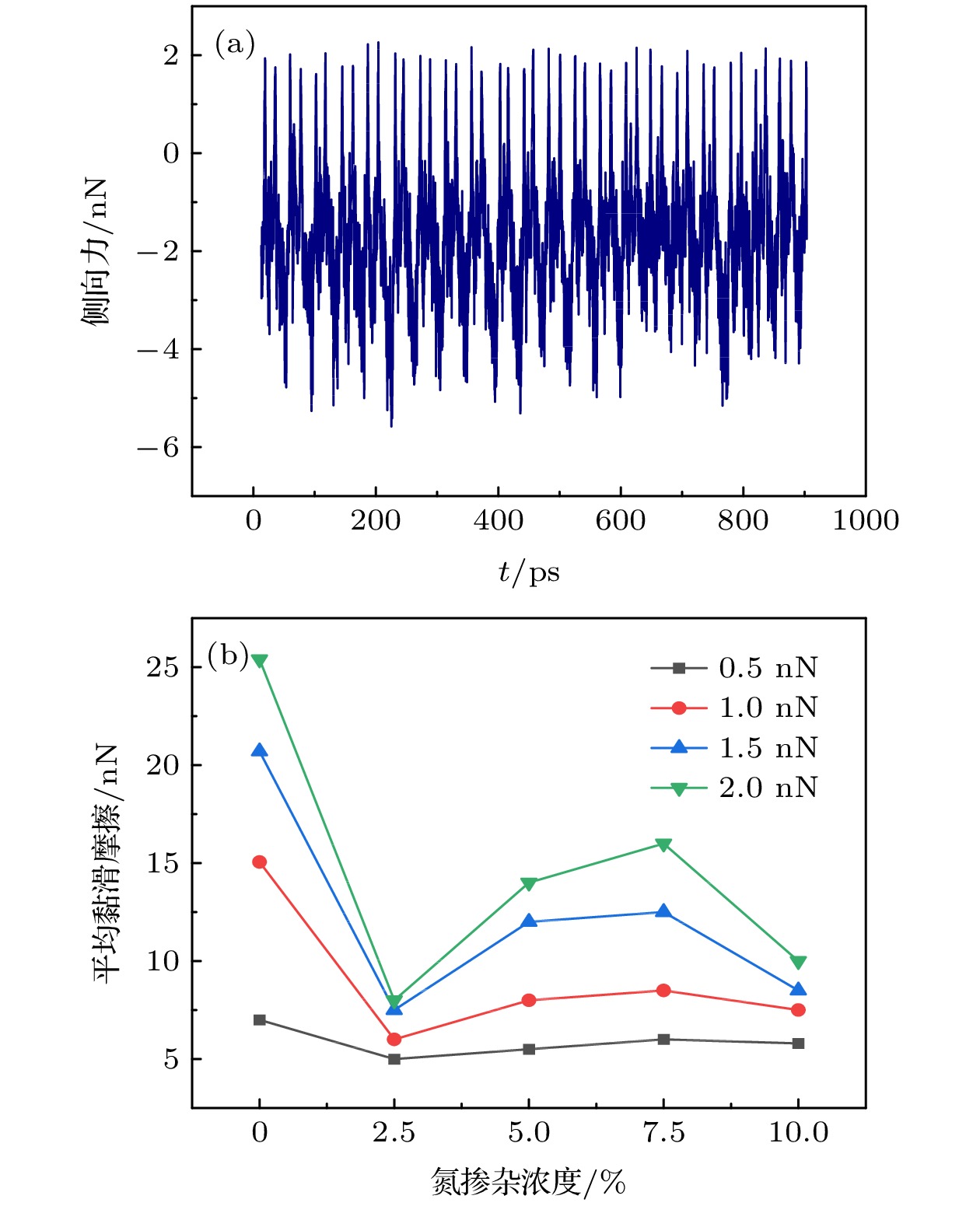
图 2 (a) 石墨烯在0.5 nN载荷下的黏滑摩擦力曲线; (b)不同载荷下界面平均摩擦力随N掺杂比例的变化
Fig. 2. (a) Viscous slip friction curve of graphene under 0.5 nN load; (b) the average friction between interfaces with nitrogen-doped (N-doped) ratio under different loads.
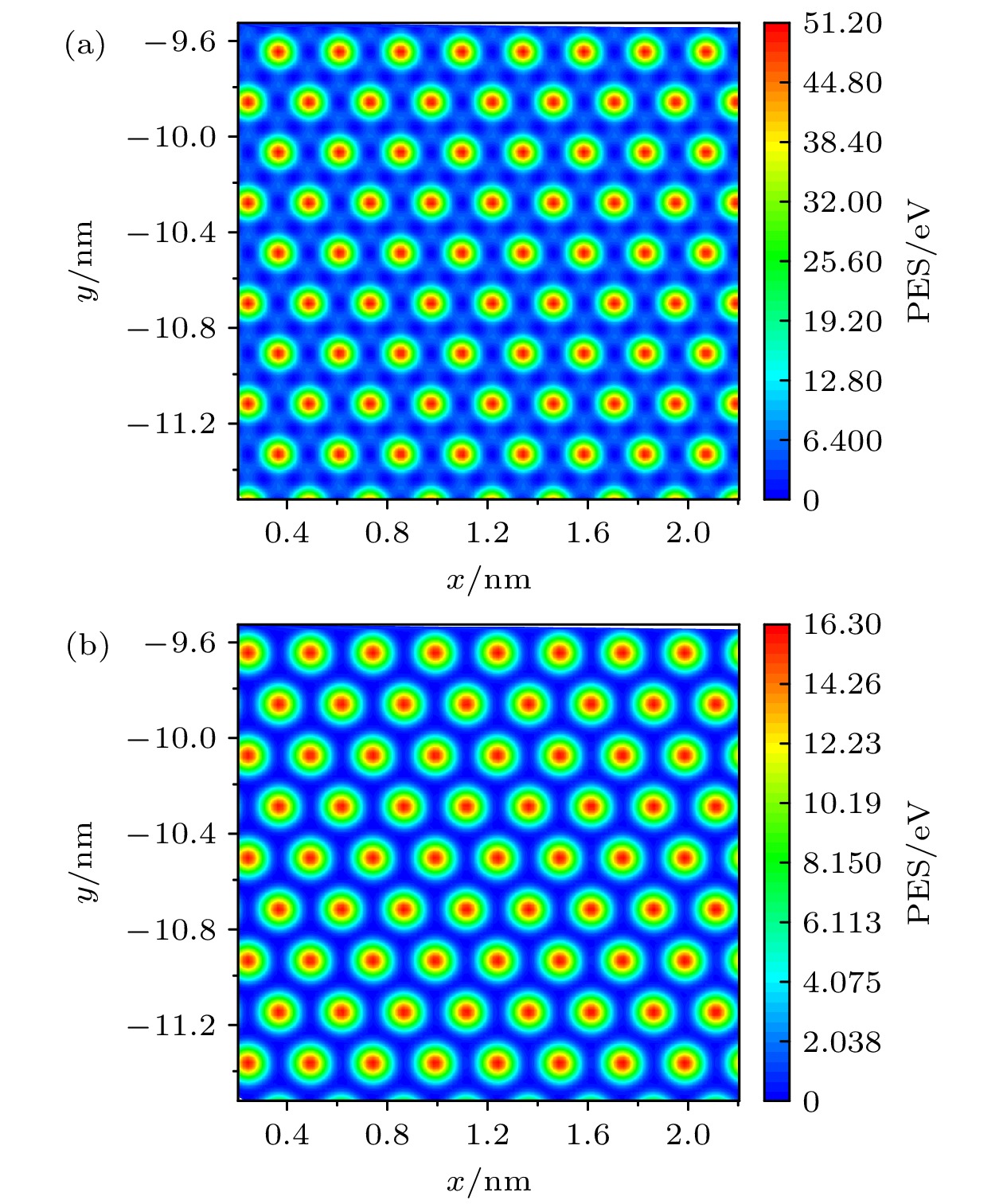
图 3 公度时两种体系界面的PES (a) 基底为石墨烯; (b) 基底为氮掺杂石墨烯
Fig. 3. PES between interfaces of two simulation systems in a commensurate state: (a) The substrate is graphene; (b) the substrate is N-doped graphene.

图 4 (a) 氮掺杂石墨烯的原子结构; (b) 理想石墨烯的原子结构
Fig. 4. (a) Atomic structure of N-doped graphene; (b) atomic structure of ideal graphene.

图 5 (a) 非公度时不同载荷下石墨烯的平均摩擦力随氮掺杂比例的变化; (b) 存在5%空位缺陷的石墨烯与N掺杂石墨烯的平均摩擦力
Fig. 5. (a) Average friction on N-doped graphene with different N-doping ratio under different loads when interface is in incommensurate state; (b) average friction on graphene with 5% vacancy defects compared with N-doped graphene.
-
[1] Novoselov K S, Geim A K, Morozov S V, Jiang D, Zhang Y, Dubonos S V, Grigorieva I V, Firsov A A 2004 Science 306 666
Google Scholar
[2] Geim A K, Novoselov K S 2007 Nat. Mater. 6 183
Google Scholar
[3] Geim A K 2009 Science 324 1530
Google Scholar
[4] Martins T B, Miwa R H, da Silva A J R, Fazzio A 2007 Phys. Rev. Lett. 98 196803
Google Scholar
[5] Dutta S, Manna A K, Pati S K 2009 Phys. Rev. Lett. 102 096601
Google Scholar
[6] Biel B, Blase X, Triozon F, Roche S 2009 Phys. Rev. Lett. 102 096803
Google Scholar
[7] Kamal Kandezi M, Shadman Lakmehsari M, Matta C F 2020 J. Mol. Liq. 302 112574
Google Scholar
[8] 林婷婷, 吕秋丰 2015 功能材料 46 5007
Google Scholar
Lin T T, Lv Q F 2015 J. Func. Mater 46 5007
Google Scholar
[9] Wang G, Wang B, Wang X, Park J, Dou S, Ahn H, Kim K 2009 J. Mate. Chem. 19 8378
Google Scholar
[10] Kwon O S, Park S J, Hong J-Y, Han A R, Lee J S, Lee J S, Oh J H, Jang J 2012 ACS. Nano. 6 1486
Google Scholar
[11] Dienwiebel M, Verhoeven G S, Pradeep N, et al. 2004 Phys. Rev. Lett. 92 126101
Google Scholar
[12] Zheng X, Gao L, Yao Q, et al. 2016 Nat. Commun. 7 13204
Google Scholar
[13] Leven I, Krepel D, Shemesh O, Hod O 2013 J. Phys. Chem. Lett. 4 115
Google Scholar
[14] Liu S W, Wang H P, Xu Q, et al. 2017 Nat. Commun. 8 14029
Google Scholar
[15] 陈勇, 李瑞 2019 物理学报 68 186801
Google Scholar
Chen Y, Li R 2019 Acta Phys. Sin. 68 186801
Google Scholar
[16] Wang K, Ouyang W, Cao W, Ma M, Zheng Q 2019 Nanoscale 11 2186
Google Scholar
[17] Liu Y, Grey F, Zheng Q 2014 Sci. Rep. 4 4875
Google Scholar
[18] Biswas S K 2008 Tribol Lett 30 159
Google Scholar
[19] Lee C, Li Q, Kalb W, Liu X Z, Berger H, Carpick R W, Hone J 2010 Science 328 76
Google Scholar
[20] Li S, Li Q, Carpick R W, Gumbsch P, Liu X Z, Ding X, Sun J, Li J 2016 Nature 539 541
Google Scholar
[21] Ye Z, Martini A 2014 Langmuir 30 14707
Google Scholar
[22] Guo Y, Guo W, Chen C 2007 Phys. Rev. B 76 155429
Google Scholar
[23] Dong Y, Wu X, Martini A 2013 Nanotechnology 24 375701
Google Scholar
[24] Gao L, Chen X, Yan Y, Liu Y, Song A, Ma T, Hu Y, Su Y, Qiao L 2018 Comput. Mater. Sci. 147 28
Google Scholar
[25] Li R, Song C 2018 Crystals 8 167
Google Scholar
[26] Shen Y, Wang Y, Zhou Y, Shi A, Hu J, Zhang Y 2016 J. Phys. D 49 415303
Google Scholar
[27] Stuart S J, Tutein A B, Harrison J A 2000 J. Chem. Phys. 112 6472
Google Scholar
[28] Kınacı A, Haskins J B, Sevik C, Çağın T 2012 Phys. Rev. B 86 115410
Google Scholar
[29] Lennard-Jones J E 1931 Proceed. Phys. Soc. 43 461
Google Scholar
[30] Leven I, Maaravi T, Azuri I, Kronik L, Hod O 2016 J. Chem. Theory Comput. 12 2896
Google Scholar
[31] Zhang W, Lu W C, Zhang H X, Ho K M, Wang C Z 2016 Carbon 110 330
Google Scholar

 下载:
下载:

计量
- 文章访问数: 8601
- PDF下载量: 158
- 被引次数: 0
