










-
利用密度泛函理论, 研究了双层石墨烯层间一氧化碳(CO)与氧(O)的氧化反应, 获得了双层石墨烯层间距与反应能垒的定量关系. 计算结果表明反应初态、过渡态、末态体系总能以及反应能垒对层间距离变化敏感: 随着层间距的逐渐缩小, 反应能垒逐渐增加. 因此, 改变双层石墨烯层间间距可以实现反应能垒的原子级调控. 通过差分电荷密度分析体系的电子结构, 发现当双层石墨烯层间距较小时, 过渡态O—C=O中碳原子与石墨烯上下层中的碳原子之间有明显的电荷堆积, 出现sp轨道杂化, 导致二者相互作用增强, 在z轴方向受到束缚力, 难以与吸附在石墨烯表面的氧原子形成较弱的O—C键, 阻碍了过渡态O—C=O的形成. 通过调控双层石墨烯间距, 可以降低一氧化碳氧化反应能垒. 该研究可为石墨烯的应用以及新型碳基插层复合材料的制备提供一定的理论支撑.Graphene is a two-dimensional (2D) crystal of carbon atoms packed in a honeycomb lattice. Because of this unique structure, it shows a number of intriguing properties. Interface between neighboring 2D layers or between 2D overlayers and substrate surfaces provides confined space for chemical process. The interlayer spacing between bilayer graphenes of van der Waals material is expected to modify the properties of atoms and molecules confined at the atomic interfaces. In this paper, the carbon monoxide (CO) and oxygen (O) in bilayer graphene are studied by density functional theory (DFT). The quantitative relationship between the interlayer spacing of bilayer graphene (d) and the reaction energy barrier (
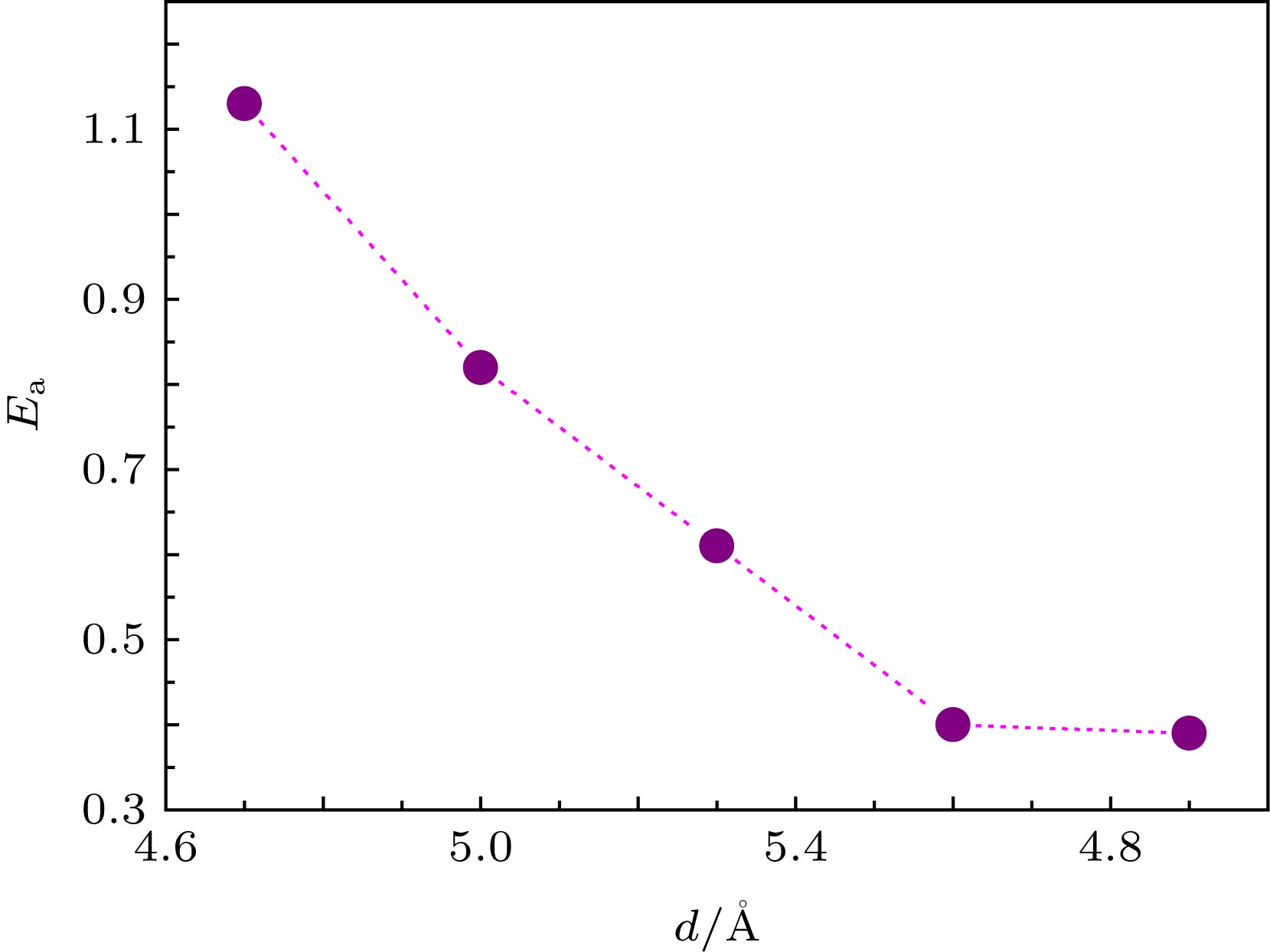
$ {{E_{\rm{a}}}} $ ) is obtained. Five values of d between 4.7 Å and 5.9 Å are used. The calculated results show that the total energy of the initial state, the transition state, the final state system and the reaction barrier are sensitive to the variation of the interlayer distance: the reaction barrier increases gradually with interlayer distance decreasing. The calculated energy barrier is 1.13 eV when the interlayer distance is 4.7 Å, while the energy barrier is 0.39 eV when the interlayer distance is 5.9 Å. It is also found that adsorption energy between O and graphene at the top site and the bridge site increase gradually with interlayer distance decreasing. Therefore, the atomic-level regulation of the reaction barrier can be achieved by changing the interlayer spacing of bilayer graphene. The charge density difference shows that when the distance between two layers of graphene is small, there is an obvious charge accumulation between C atoms in transition state O—C=O and C atoms in the upper or lower layer of graphene. This results in sp orbital hybridization, which leads the interaction between two C atoms to be enhanced. It is difficult to form a weak O—C bond of transition state O—C=O with O atoms adsorbed on graphene because of a binding force which exists in the z-axis direction. The DFT calculation of CO oxidation reaction barrier can be reduced by adjusting the spacing of bilayer graphene, which provides a theoretical support for the application of graphene and the preparation of new carbon-based intercalated composites.-
Keywords:
- grapheme /
- confined reaction /
- density functional theory /
- reaction energy barrier
[1] Novoselov K S, Geim A K, Morozov S V, Jiang D, Zhang Y, Dubonos S V, Grigorieva I V, Firsov A A 2004 Science 306 666
 Google Scholar
Google Scholar
[2] Mao Y, Yuan J, Zhong J 2010 J. Phys. Condens. Matt. 405 3337
Google Scholar
[3] Liu X, Wang C Z, Yao Y X 2011 Phys. Rev. B 83 235411
[4] El-Kady M F, Strong V, Dubin S, Kaner R B 2012 Science 335 1326
Google Scholar
[5] Reddy A L M, Srivastava A, Gowda S R, Gullapalli H, Dubey M, Ajayan P M 2010 ACS Nano 4 6337
Google Scholar
[6] Verhoff F H, Labourt-Ibarre P, Ballal G D 1981 Chem. Eng. Sci. 36 1713
[7] Yao Y X, Fu Q, Zhang Y Y, Weng X F, Li H, Chen M S, Jin L, Dong A Y, Mu R T, Jiang P, Liu L, Bluhm H, Liu Z, Zhang S B, Bao X H 2014 Proc. Natl. Acad. Sci. USA 111 17023
Google Scholar
[8] Fu Q, Bao X 2017 Chem. Soc. Rev. 46 1842
Google Scholar
[9] Zhang Y, Wang X, Li H, Li H, Wei M, Xiao X, Liu Z, Chen M, Fu Q, Bao X 2015 Nano Lett. 15 3616
Google Scholar
[10] Deng, D, Novoselov K, Fu Q, Zheng N, Tian Z, Bao X 2016 Nat. Nanotechnol. 11 218
Google Scholar
[11] Sutter P, Sadowski J T, Sutter E A 2010 J. Am. Chem. Soc. 132 8175
Google Scholar
[12] Ferrighi L, Datteo M, Fazio G, Di Valentin C 2016 J. Am. Chem. Soc. 138 7365
Google Scholar
[13] Lei F, Liu W, Sun Y, Xu J, Liu K, Liang L, Yao T, Pan B, Wei S, Xie Y 2016 Nat. Commun. 7 12697
Google Scholar
[14] Zhang H, Fu Q, Cui Y, Tan D, Bao X 2009 J. Phys. Chem. C 113 8296
[15] Li H, Xiao J, Fu Q, Bao X 2017 Proc. Natl. Acad. Sci. USA 114 5930
[16] Wang W X, Wei Y W, Li S Y, Li X Q, Wu X S, Feng J, He L 2018 Phys. Rev. B 97 085407
Google Scholar
[17] 周晓峰, 方浩宇, 唐春梅. 2019 物理学报 68 053601
Google Scholar
Zhou X F, Fang H Y, Tang C M 2019 Acta Phys. Sin. 68 053601
Google Scholar
[18] Kresse G, Furthmuller J 1996 Phys. Rev. B 54 11169
Google Scholar
[19] Kresse G, Furthmuller J 1996 Comput. Mater. Sci. 6 15
Google Scholar
[20] Perdew J P, Burke K, Ernzerhof M 1996 Phys. Rev. Lett. 77 3865
Google Scholar
[21] Blochl P E 1994 Phys. Rev. B 50 17953
Google Scholar
[22] Kresse G, Joubert D 1999 Phys. Rev. B 59 1758
[23] Grimme S, Antony J, Ehrlich S, Krieg H 2010 J. Chem. Phys. 132 154104
Google Scholar
[24] Henkelman G, Uberuaga B P, Jónsson H 2000 J. Chem. Phys. 113 9901
Google Scholar
[25] Henkelman G, Jónsson H 2000 J. Chem. Phys. 113 9978
Google Scholar
[26] Liu X, Sui Y H, Duan T, Meng C G, Han Y 2014 Phys. Chem. Chem. Phys. 16 23584
Google Scholar
-
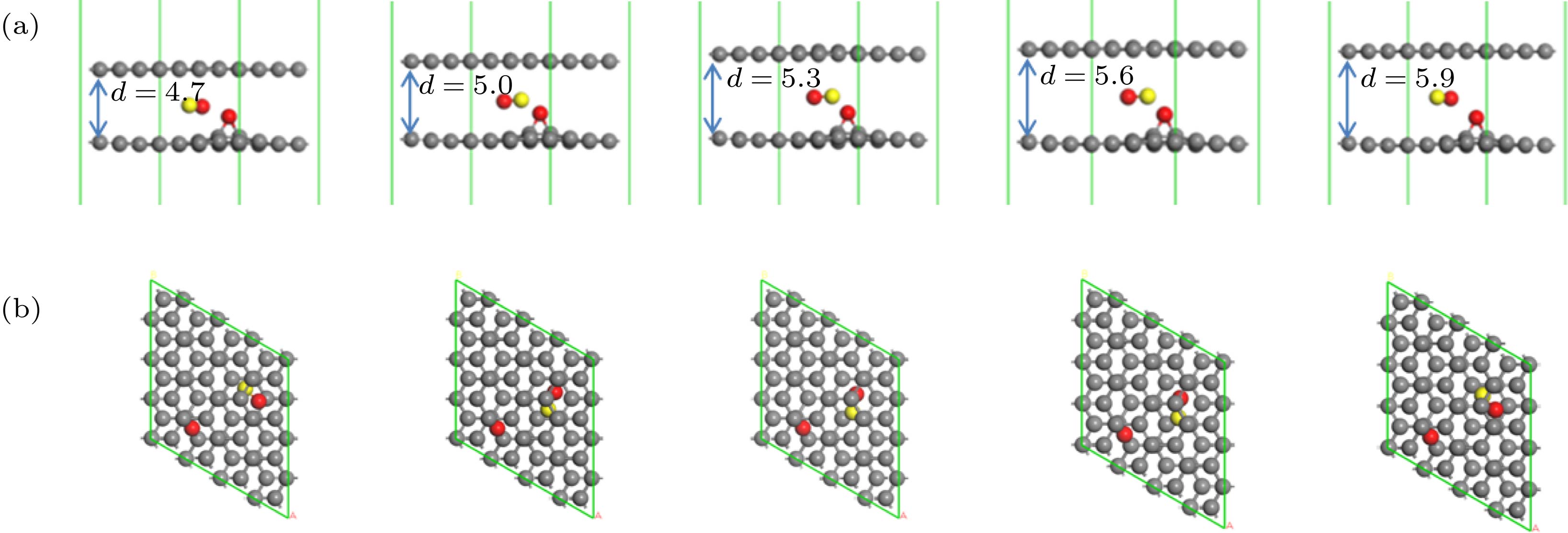
图 1 初始结构优化模型 (a)侧面图; (b)俯视图
Fig. 1. Side view (a) and top view (b) of the initial optimized structures about different models.

图 3 各状态对应结构的侧视图 (a)初态; (b)过渡态; (c)末态
Fig. 3. Side view of local configurations at various states along the reaction pathway: (a) Initial state; (b) transition state; (c) final state.

图 4 不同层间距下, 从初态到末态一氧化碳氧化反应路径 (a) d = 4.7 Å; (b) d = 5.0 Å; (c) d = 5.3 Å; (d) d = 5.6 Å; (e) d = 5.9 Å
Fig. 4. Reaction pathway of the oxidation of CO and O from the initial state to final state under different interlayer distance: (a) d = 4.7 Å, (b) d = 5.0 Å, (c) d = 5.3 Å, (d) d = 5.6 Å, (e) d = 5.9 Å.
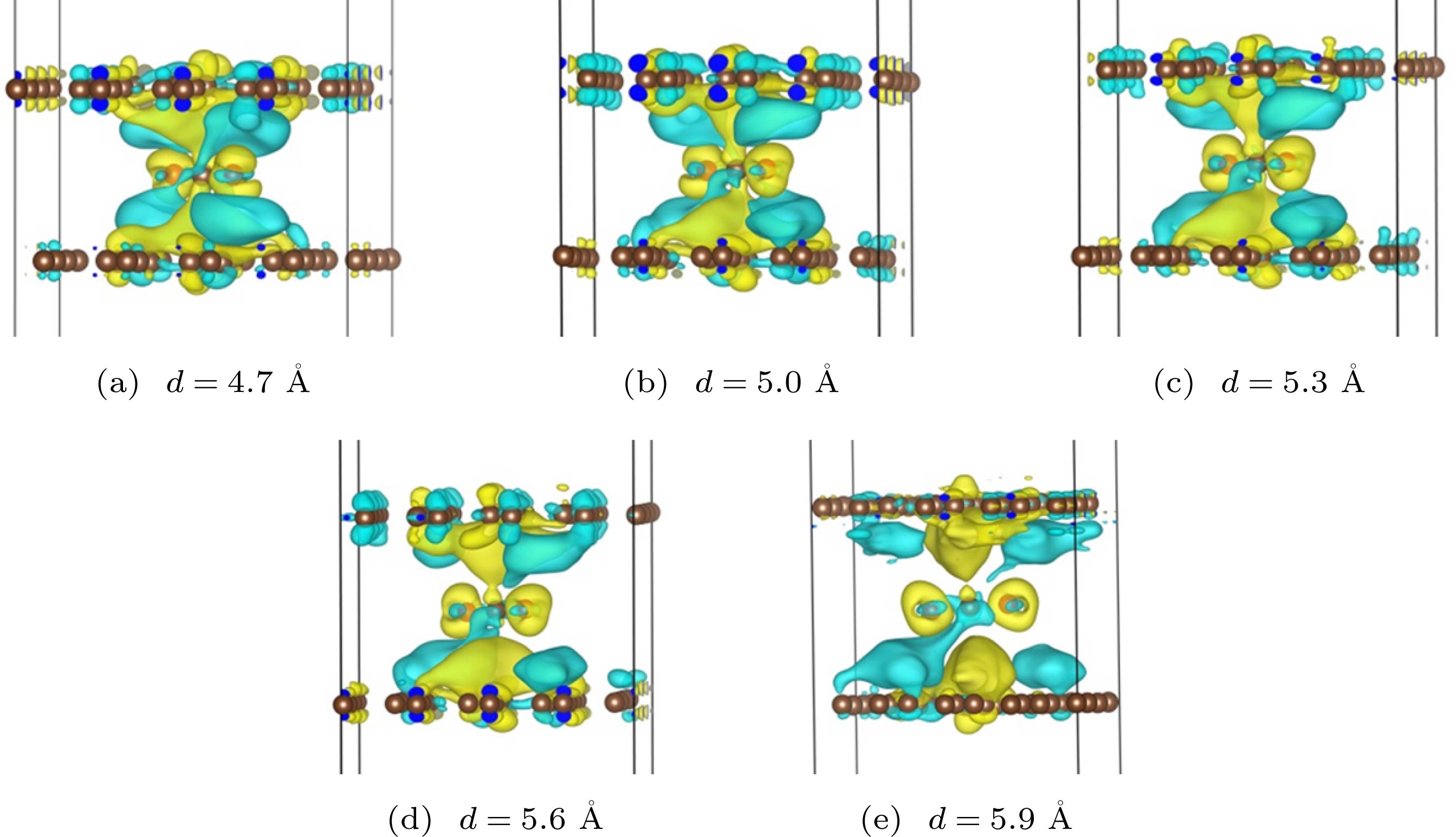
图 5 CO与O反应, 在不同间距的双层石墨烯层间过渡态的电荷差分密度图, 其中等值面数值分别为 (a) 0.00014 e (a.u.)3; (b) 0.00020 e (a.u.)3; (c) 0.00025 e (a.u.)3; (d) 0.00035 e (a.u.)3; (e) 0.00048 e (a.u.)3
Fig. 5. Electron density different of the transition states between the bilayer grapheme and the isosurface is (a) 0.00014 e (a.u.)3, (b) 0.00020 e (a.u.)3, (c) 0.00025 e (a.u.)3, (d) 0.00035 e (a.u.)3, (e) 0.00048 e (a.u.)3, respectively.
表 1 五个不同间距情况下的
$E_{{\rm{ads}}{\text{-}}{\rm{O}}}^{{\rm{Top}}}({\rm{IS}})$ ,$E_{{\rm{ads}}{\text{-}}{\rm{O}}}^{{\rm{Bridge}}}({\rm{IS}})$ ,${E_{\rm{a}}}$ 和$\Delta H$ Table 1. Adsorption energy at top site and bridge site, the reaction energy barrier and reaction heat at five different interlayer distances.
Model d $\small E_{ {\rm{ads} }{\text{-} }{\rm{O} } }^{ {\rm{Top} } }({\rm{IS} })$ $\small E_{ {\rm{ads} } {\text{-} }{\rm{O} } }^{ {\rm{Bridge} } }({\rm{IS} })$ ${E_{\rm{a}}}$ $\Delta H$ 1 4.7 –1.85 –2.51 1.13 –3.82 2 5.0 –1.81 –2.47 0.82 –3.97 3 5.3 –1.68 –2.34 0.61 –4.05 4 5.6 –1.41 –2.07 0.40 –4.08 5 5.9 –0.90 –1.56 0.39 –4.10 注: d 是双层石墨烯层间距.  下载: 导出CSV
下载: 导出CSV
表 2 五个不同间距时初态(IS)、过渡态(TS)、末态(FS)的CO中C—O键长, O与CO分子间距以及CO2 (O=C=O)中O—C与C—O键长(分别对应dC—O(CO), dCO—O, dO—C(CO2), dC—O(CO2), 单位为Å)
Table 2. The C—O bond lengths of the initial, transition and final states of CO at five different distances, the molecular distances between O and CO, and the O—C and C—O bond lengths in CO2 (O=C=O). They correspond to dC—O(CO), dCO—O, dO—C(CO2), dC—O(CO2) with the units of Å.
Model Reaction state IS TS FS dC—O(CO) dCO—O dC—O(CO) dCO—O dO—C(CO2) dC—O(CO2) d = 4.7 1.153 4.170 1.170 2.057 1.175 1.175 d = 5.0 1.149 3.463 1.172 2.118 1.175 1.175 d = 5.3 1.148 3.385 1.168 2.110 1.176 1.176 d = 5.6 1.146 3.530 1.163 2.038 1.176 1.176 d = 5.7 1.145 4.338 1.167 2.276 1.176 1.176
下载: 导出CSV
-
[1] Novoselov K S, Geim A K, Morozov S V, Jiang D, Zhang Y, Dubonos S V, Grigorieva I V, Firsov A A 2004 Science 306 666
Google Scholar
[2] Mao Y, Yuan J, Zhong J 2010 J. Phys. Condens. Matt. 405 3337
Google Scholar
[3] Liu X, Wang C Z, Yao Y X 2011 Phys. Rev. B 83 235411
[4] El-Kady M F, Strong V, Dubin S, Kaner R B 2012 Science 335 1326
Google Scholar
[5] Reddy A L M, Srivastava A, Gowda S R, Gullapalli H, Dubey M, Ajayan P M 2010 ACS Nano 4 6337
Google Scholar
[6] Verhoff F H, Labourt-Ibarre P, Ballal G D 1981 Chem. Eng. Sci. 36 1713
[7] Yao Y X, Fu Q, Zhang Y Y, Weng X F, Li H, Chen M S, Jin L, Dong A Y, Mu R T, Jiang P, Liu L, Bluhm H, Liu Z, Zhang S B, Bao X H 2014 Proc. Natl. Acad. Sci. USA 111 17023
Google Scholar
[8] Fu Q, Bao X 2017 Chem. Soc. Rev. 46 1842
Google Scholar
[9] Zhang Y, Wang X, Li H, Li H, Wei M, Xiao X, Liu Z, Chen M, Fu Q, Bao X 2015 Nano Lett. 15 3616
Google Scholar
[10] Deng, D, Novoselov K, Fu Q, Zheng N, Tian Z, Bao X 2016 Nat. Nanotechnol. 11 218
Google Scholar
[11] Sutter P, Sadowski J T, Sutter E A 2010 J. Am. Chem. Soc. 132 8175
Google Scholar
[12] Ferrighi L, Datteo M, Fazio G, Di Valentin C 2016 J. Am. Chem. Soc. 138 7365
Google Scholar
[13] Lei F, Liu W, Sun Y, Xu J, Liu K, Liang L, Yao T, Pan B, Wei S, Xie Y 2016 Nat. Commun. 7 12697
Google Scholar
[14] Zhang H, Fu Q, Cui Y, Tan D, Bao X 2009 J. Phys. Chem. C 113 8296
[15] Li H, Xiao J, Fu Q, Bao X 2017 Proc. Natl. Acad. Sci. USA 114 5930
[16] Wang W X, Wei Y W, Li S Y, Li X Q, Wu X S, Feng J, He L 2018 Phys. Rev. B 97 085407
Google Scholar
[17] 周晓峰, 方浩宇, 唐春梅. 2019 物理学报 68 053601
Google Scholar
Zhou X F, Fang H Y, Tang C M 2019 Acta Phys. Sin. 68 053601
Google Scholar
[18] Kresse G, Furthmuller J 1996 Phys. Rev. B 54 11169
Google Scholar
[19] Kresse G, Furthmuller J 1996 Comput. Mater. Sci. 6 15
Google Scholar
[20] Perdew J P, Burke K, Ernzerhof M 1996 Phys. Rev. Lett. 77 3865
Google Scholar
[21] Blochl P E 1994 Phys. Rev. B 50 17953
Google Scholar
[22] Kresse G, Joubert D 1999 Phys. Rev. B 59 1758
[23] Grimme S, Antony J, Ehrlich S, Krieg H 2010 J. Chem. Phys. 132 154104
Google Scholar
[24] Henkelman G, Uberuaga B P, Jónsson H 2000 J. Chem. Phys. 113 9901
Google Scholar
[25] Henkelman G, Jónsson H 2000 J. Chem. Phys. 113 9978
Google Scholar
[26] Liu X, Sui Y H, Duan T, Meng C G, Han Y 2014 Phys. Chem. Chem. Phys. 16 23584
Google Scholar


 下载:
下载:






计量
- 文章访问数: 13145
- PDF下载量: 122
- 被引次数: 0
